HI-VQE Chemistry – Eine Qiskit Function von Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Siehe die API-Referenz
Qiskit Functions sind ein experimentelles Feature, das ausschließlich Nutzerinnen und Nutzern des IBM Quantum® Premium Plan, Flex Plan und On-Prem (über die IBM Quantum Platform API) Plan zur Verfügung steht. Sie befinden sich im Preview-Release-Status und können sich noch ändern.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
Überblick
In der Quantenchemie befasst sich das Problem der Elektronenstruktur mit der Lösung der elektronischen Schrödinger-Gleichung – also der Quantenwellenfunktionen, die das Verhalten der Elektronen im System beschreiben. Diese Wellenfunktionen sind Vektoren komplexer Amplituden, wobei jede Amplitude dem Beitrag einer möglichen Elektronenkonfiguration entspricht.
Der Grundzustand ist die Wellenfunktion niedrigster Energie des Systems und hat bei der Untersuchung molekularer Systeme eine besondere Bedeutung. Der genaueste Ansatz zur Berechnung des Grundzustands berücksichtigt alle möglichen Elektronenkonfigurationen, wird aber für größere Systeme schnell unlösbar, da die Anzahl der Konfigurationen exponentiell mit der Systemgröße wächst.
Der Handover Iterative Variational Quantum Eigensolver (HI-VQE) ist eine innovative hybride Quanten-klassische Methode zur genauen Schätzung des Grundzustands molekularer Systeme. Er verbindet Quantenhardware mit klassischem Computing: Quantenprozessoren erkunden effizient Kandidaten-Elektronenkonfigurationen, während die resultierende Wellenfunktion auf klassischen Rechnern berechnet wird. Durch die Erzeugung kompakter, aber chemisch genauer Wellenfunktionen fördert HI-VQE Forschung und Entdeckungen in der Quantenchemie und Materialwissenschaft.

HI-VQE reduziert die Rechenkomplexität des Elektronenstrukturproblems, indem es den Grundzustand effizient und mit hoher Genauigkeit schätzt. Der Fokus liegt auf einer sorgfältig ausgewählten Teilmenge der relevantesten Elektronenkonfigurationen, was sowohl Genauigkeit als auch Effizienz optimiert.
Durch die Kombination der Stärken klassischer und Quantenrechner verfeinert HI-VQE iterativ die aktuelle Schätzung der Wellenfunktion. Seine einzigartigen Techniken zur Teilraumkonstruktion machen die Konfigurationsauswahl effizienter, sodass Nutzerinnen und Nutzer mehr Rechenkontrolle und verbesserte Genauigkeit in Quantenchemiesimulationen erhalten.
Wenn du den Algorithmus eingehender kennenlernen möchtest, kannst du das zugehörige Forschungspapier lesen
Beschreibung
Die Anzahl der Elektronenkonfigurationen eines molekularen Systems wächst exponentiell mit der Systemgröße. Bei bestimmten elektronischen Zuständen, wie dem Grundzustand, tragen jedoch häufig nur wenige Konfigurationen wesentlich zur Energie des Zustands bei. Methoden der ausgewählten Konfigurationswechselwirkung (Selected Configuration Interaction, SCI) nutzen diese Spärlichkeit, um Rechenkosten zu senken, indem sie die relevantesten Konfigurationen identifizieren und auf sie fokussieren. Diese Teilmenge der Konfigurationen wird als Teilraum (Subspace) bezeichnet.
HI-VQE nutzt die inhärente Effizienz von Quantencomputern bei der Darstellung molekularer Systeme, um die Teilraumsuche zu unterstützen. Es verbindet klassische und Quanten-Subroutinen, um das Elektronenstrukturproblem mit hoher Genauigkeit zu lösen. Im Gegensatz zu bestehenden Quanten-SCI-Methoden kombiniert HI-VQE Variationstraining, iterative Teilraumkonstruktion und Vordiagonalisierungs-Konfigurations-Screening, um die Effizienz durch Reduzierung von Quantenmessungen, Iterationen und klassischen Diagonalisierungskosten zu steigern. HI-VQE kann daher auf größere molekulare Systeme angewendet werden, die mehr Qubits benötigen, und senkt die Kosten zur Lösung eines Problems gegebener Größe auf denselben Genauigkeitsgrad.
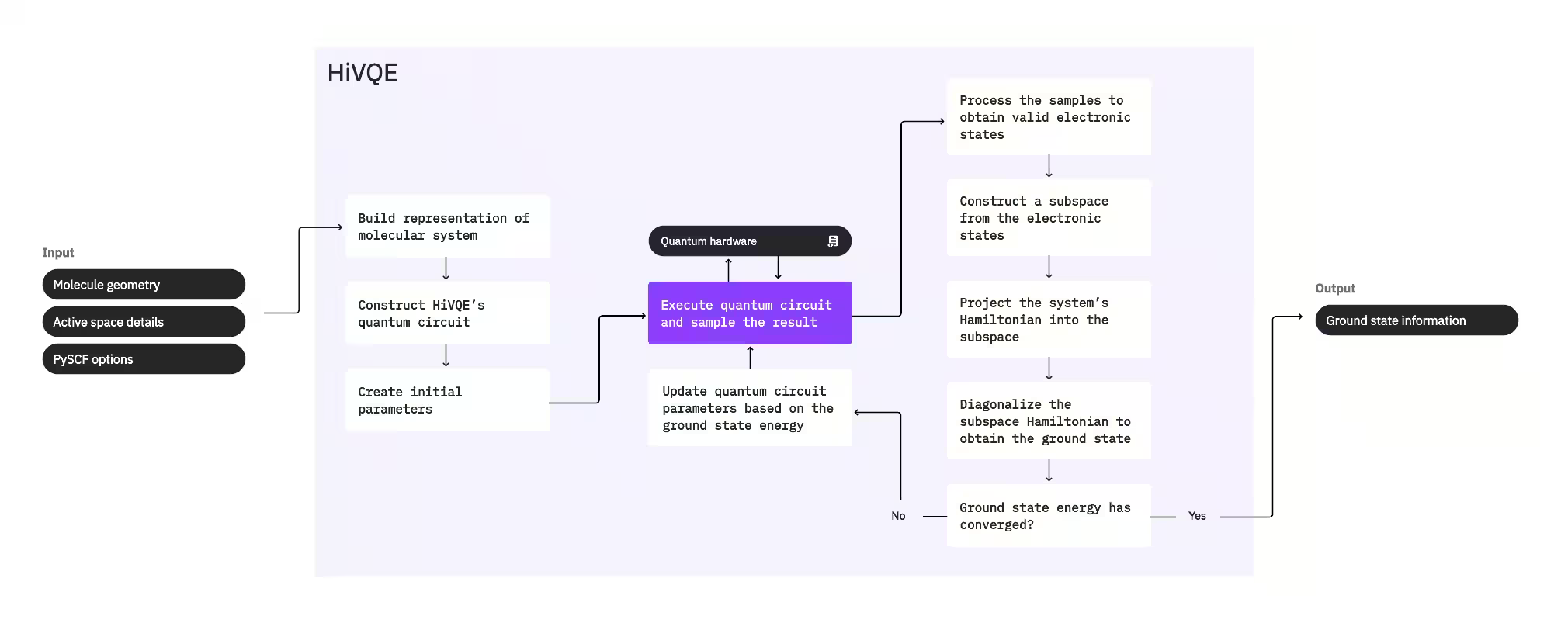
Um den Grundzustand eines Systems zu berechnen, verwendet HI-VQE zunächst das klassische Chemiepaket PySCF, um aus nutzergegebenen Eingaben – wie der Molekülgeometrie und weiteren molekularen Informationen – eine molekulare Darstellung zu erzeugen. Anschließend tritt es in eine hybride Quanten-klassische Optimierungsschleife ein, die iterativ einen Teilraum verfeinert, um den Grundzustand optimal darzustellen und gleichzeitig die Anzahl der enthaltenen Konfigurationen zu minimieren. Die Schleife läuft bis Konvergenzkriterien – wie Teilraumgröße oder Energiestabilität – erfüllt sind; danach werden die berechnete Grundzustandswellenfunktion und Energie ausgegeben. Diese Ergebnisse können genutzt werden, um genaue Potentialenergieflächen zu konstruieren und weitere Analysen des Systems durchzuführen.
Die Optimierungsschleife konzentriert sich darauf, die Parameter eines Quantum Circuits anzupassen, um einen hochwertigen Teilraum zu erzeugen. HI-VQE bietet drei Quantum Circuit-Optionen: excitation_preserving, efficient_su2 und LUCJ. Die Optimierung wird nahe dem Hartree-Fock-Referenzzustand initialisiert, da dieser sich allgemein gut eignet. Der Circuit wird dann auf einem Quantengerät ausgeführt und Konfigurationen werden aus dem resultierenden Quantenzustand gesampelt, bevor sie als binäre Zeichenketten zurückgegeben werden. Aufgrund von Quantengeräterauschen können einige gesampelte Konfigurationen physikalisch ungültig sein und die Elektronenzahl oder den Spin nicht erhalten. HI-VQE begegnet diesem Problem mit dem Konfigurationswiederherstellungsprozess aus dem qiskit-addon-sqd-Paket, sodass Nutzerinnen und Nutzer ungültige Konfigurationen entweder korrigieren oder verwerfen können.
Die gültigen Konfigurationen durchlaufen dann einen optionalen Screening-Schritt, um jene zu entfernen, die voraussichtlich nur minimal beitragen. Dies reduziert die Dimension des Teilraums und senkt dadurch die Kosten des Diagonalisierungsschritts. Wenn das Screening aktiviert ist, wird ein vorläufiger Teilraum-Hamiltonoperator aus den gültigen Konfigurationen aufgebaut und eine Diagonalisierung mit sehr lockeren Abbruchkriterien durchgeführt. Obwohl die Genauigkeit der resultierenden Amplituden für jede Konfiguration gering ist, eignet sie sich gut dafür vorherzusagen, welche Konfigurationen in dieser Iteration aus dem Teilraum herausgelassen werden sollen – und sie ist schnell zu berechnen.
Die ausgewählten Konfigurationen werden dem Teilraum hinzugefügt und der Hamiltonoperator des Systems in diesen Teilraum projiziert. Der Teilraum wird iterativ aktualisiert und behält dabei die relevantesten Konfigurationen über Iterationen hinweg. Dieser Ansatz unterscheidet sich von alternativen Methoden, da der Quantum Circuit nicht bei jedem Schritt den vollständigen Grundzustand approximieren muss.
Anschließend wird der Teilraum-Hamiltonoperator klassisch diagonalisiert, um den niedrigsten Eigenwert und den zugehörigen Eigenvektor zu erhalten – eine Approximation des Grundzustands und seiner Energie. Da sich die Qualität des Teilraums durch die Iterationen verbessert, nähert sich der berechnete Grundzustand immer mehr dem wahren Grundzustand an. In diesem Schritt kann ein zusätzlicher Screening-Schritt durchgeführt werden, um Konfigurationen aus dem Teilraum zu entfernen, die keinen wesentlichen Beitrag zum berechneten Grundzustand leisten. Dieser Schritt stellt sicher, dass der in die nächste Iteration übergehende Teilraum so kompakt wie möglich ist. Die Bewertung erfolgt anhand der Amplituden aus der Diagonalisierung, da diese den Wichtigkeitsbeitrag jeder Konfiguration zum berechneten Grundzustand darstellen.
Eine Konvergenzprüfung stellt dann fest, ob weiteres Training die Ergebnisse verbessern würde. Falls ja, wird ein optionaler klassischer Erweiterungsschritt durchgeführt, die Parameter des Quantum Circuits werden aktualisiert, um die berechnete Energie weiter zu minimieren, und der Prozess wiederholt sich. Der klassische Erweiterungsschritt erzeugt zusätzliche Konfigurationen für den Teilraum und ergänzt damit die vom Quantengerät gesampelten Konfigurationen. Dabei wird zunächst die Konfiguration mit der größten Amplitude in den Diagonalisierungsergebnissen identifiziert, bevor neue Konfigurationen mit einfachen und doppelten Anregungen aus der identifizierten Konfiguration generiert werden. Die gewünschte Anzahl dieser Konfigurationen wird dann dem Teilraum hinzugefügt.
Sobald festgestellt wird, dass die Iterationen konvergiert sind, gibt HI-VQE den berechneten Grundzustand (in Form der Zustände im Teilraum und ihrer Amplituden in der Grundzustandswellenfunktion), seine Energie und ein Energievarianzmaß zurück, das angibt, ob der berechnete Zustand einen Eigenzustand des Hamiltonoperators des Systems bildet.
Nutzerinnen und Nutzer können den verwendeten Quantum Circuit und die Anzahl der Shots pro Quantum Circuit wählen sowie die Teilraumgröße steuern oder die klassische Generierung zusätzlicher Konfigurationen aktivieren, um die quantengenerierten Konfigurationen zu ergänzen. Auf diese Weise lässt sich das Verhalten von HI-VQE an die jeweiligen Anwendungsfälle anpassen.
Lizenzierung
Bitte beachte, dass die Nutzung dieser Qiskit Function auf Probleme mit höchstens 20 Qubits beschränkt ist, sofern keine Lizenz erworben wird, die ein höheres Limit gewährt.
Bitte sende eine E-Mail an qiskit.support@qunovacomputing.com, wenn du Informationen zum Erwerb einer Lizenz einholen möchtest.
Erste Schritte
Zuerst beantrage Zugang zur Function. Authentifiziere dich dann mit deinem IBM Quantum® API-Schlüssel und – vorausgesetzt, du hast dein Konto bereits in deiner lokalen Umgebung gespeichert – wähle die Qiskit Function wie folgt aus:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Beispiel
Das erste Beispiel zeigt, wie die Grundzustandsenergie eines NH3-Moleküls mit dem HI-VQE-Algorithmus berechnet wird.
Molekülgeometrie und Optionen definieren
Die Molekülgeometrie von NH3 wird mit kartesischen Koordinaten angegeben, getrennt durch ";" für jedes Atom.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Weitere Optionen können für das Molekülsystem im folgenden Dictionary-Format definiert und übergeben werden.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Führe die Function mit den Geometrie- und Optionseingaben aus.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Es ist empfehlenswert, die Job-ID der Function auszugeben, damit sie bei Support-Anfragen angegeben werden kann, falls etwas schiefgeht.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
In diesem Beispiel werden 16 Qubits mit 8 Orbitalen der sto3g-Basis für ein NH3-Molekül verwendet. Überprüfe den Status deiner Qiskit Function-Arbeitslast oder rufe die Ergebnisse wie folgt ab:
print(job.status())
QUEUED
Nach Abschluss des Jobs können die Ergebnisse mit der result()-Instanz abgerufen werden.
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Um auf die Grundzustandsenergie zuzugreifen, verwende den Schlüssel "energy". Der Schlüssel "eigenvector" liefert die CI-Koeffizienten mit entsprechender Bitstring-Notation der Elektronenkonfiguration, die unter "states" der Ergebnisse gespeichert ist.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Leistung
Dieser Abschnitt zeigt die nachgewiesenen Benchmark-Berechnungen von HI-VQE mit einem 24-Qubit-Fall für Li2S, einem 40-Qubit-Fall für ein N2-Molekül und einem 44-Qubit-Fall für ein FeP-NO-System.
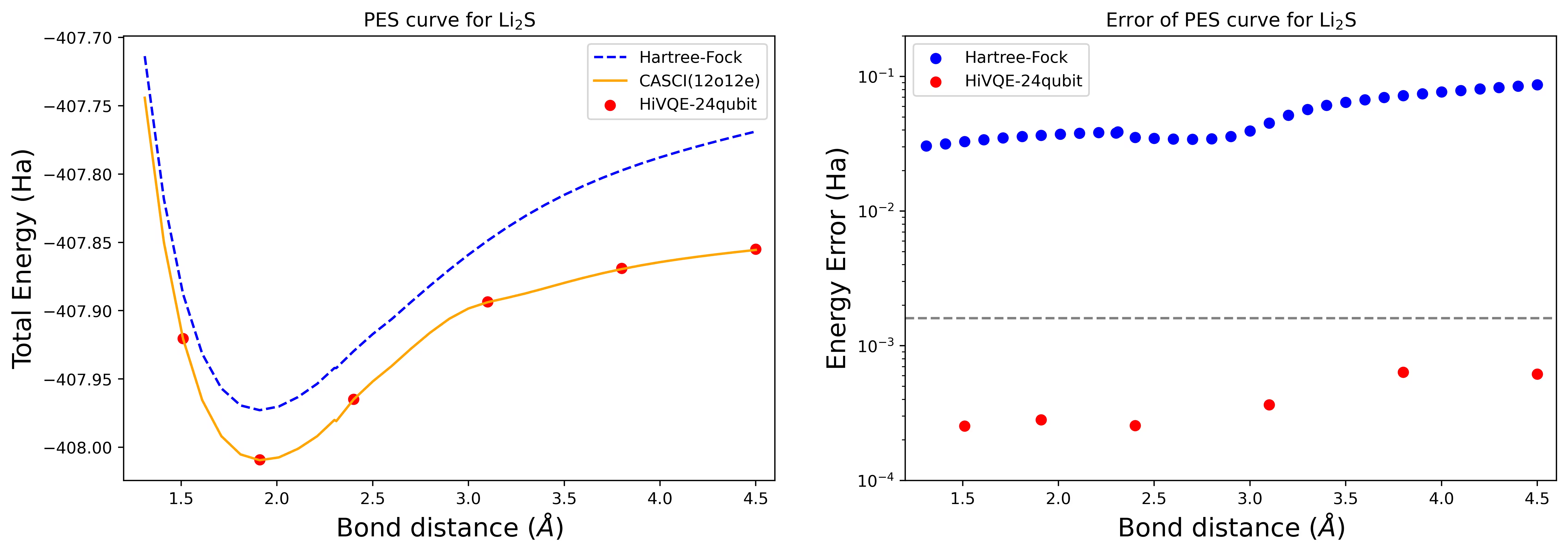
Dissoziations-Potentialenergieflächenkurve für ein Li2S-Molekül mit 24 Qubits
Die PES-Kurve wird mit der FCI-Referenz und dem Startwert aus RHF zusammen mit dem Energiefehler gegenüber der FCI-Referenz gezeigt.
 .
.
Die Berechnungen wurden mit den folgenden Geometrien und Optionen durchgeführt.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Die roten Punkte stellen die HI-VQE-Berechnungsergebnisse für sechs verschiedene Geometrien dar; drei Geometrien entsprechend 1,51, 2,40 und 3,80 Ångström sind als Eingabe in der obigen Zelle angegeben.
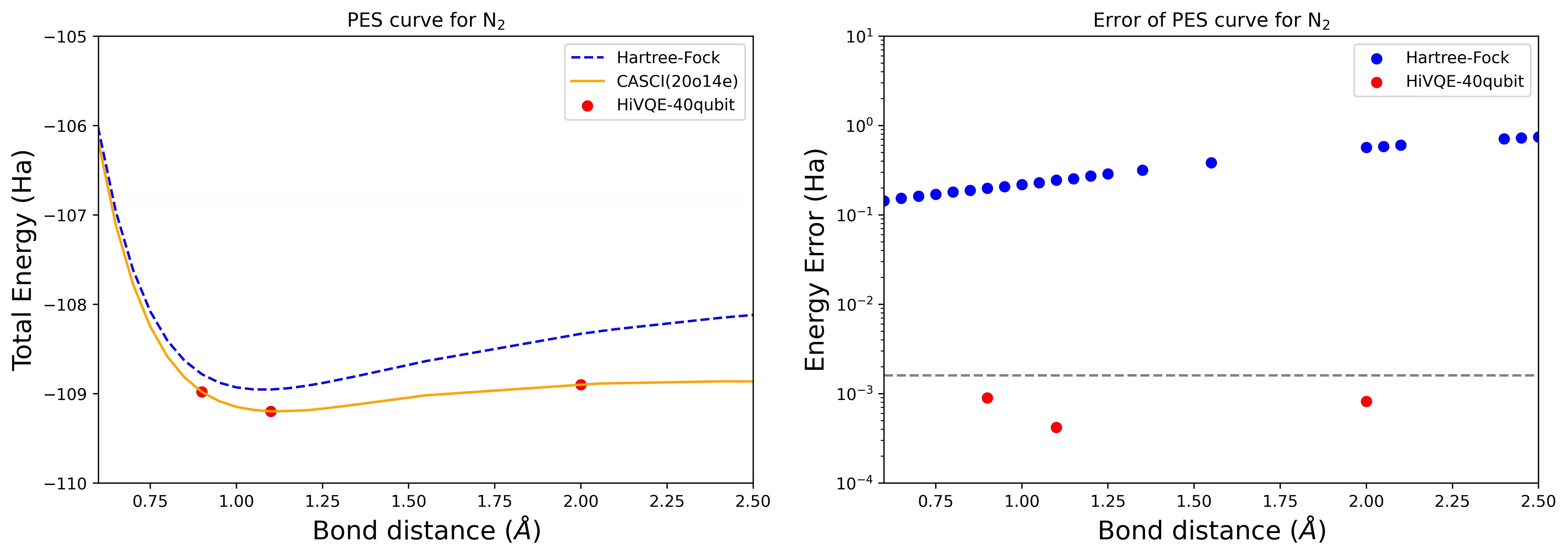
Dissoziations-PES-Kurve für ein N2-Molekül mit 40 Qubits
Das Stickstoffmolekül wurde als Multireferenzsystem mit großen Korrelationsenergiebeträgen über den Hartree-Fock-Zustand hinaus identifiziert. Wir haben eine Benchmark-Berechnung für das N2-Molekül mit cc-pvdz-Basis, (20o,14e) unter Verwendung der homo-lumo aktiven Orbitalauswahl durchgeführt. Die vollständige aktive Raumzahl (CAS), um dieses Problem darzustellen, beträgt 6.009.350.400. Es ist nicht möglich, die Eigenwertaufgabe (für Energie und Elektronenstruktur) mit dieser Anzahl von Zuständen auf einem leistungsstarken Desktop-Rechner (16 CPU/64 GB) zu lösen. Mit HI-VQE können Nutzerinnen und Nutzer den Teilraum der CAS-Zustände effizient durchsuchen, um chemisch genaue Ergebnisse zu erzielen und dabei Rechenressourcen erheblich einzusparen. Die folgenden Diagramme zeigen die PES-Kurve der 40-Qubit-HI-VQE-Berechnung der N2-Moleküldissoziation.
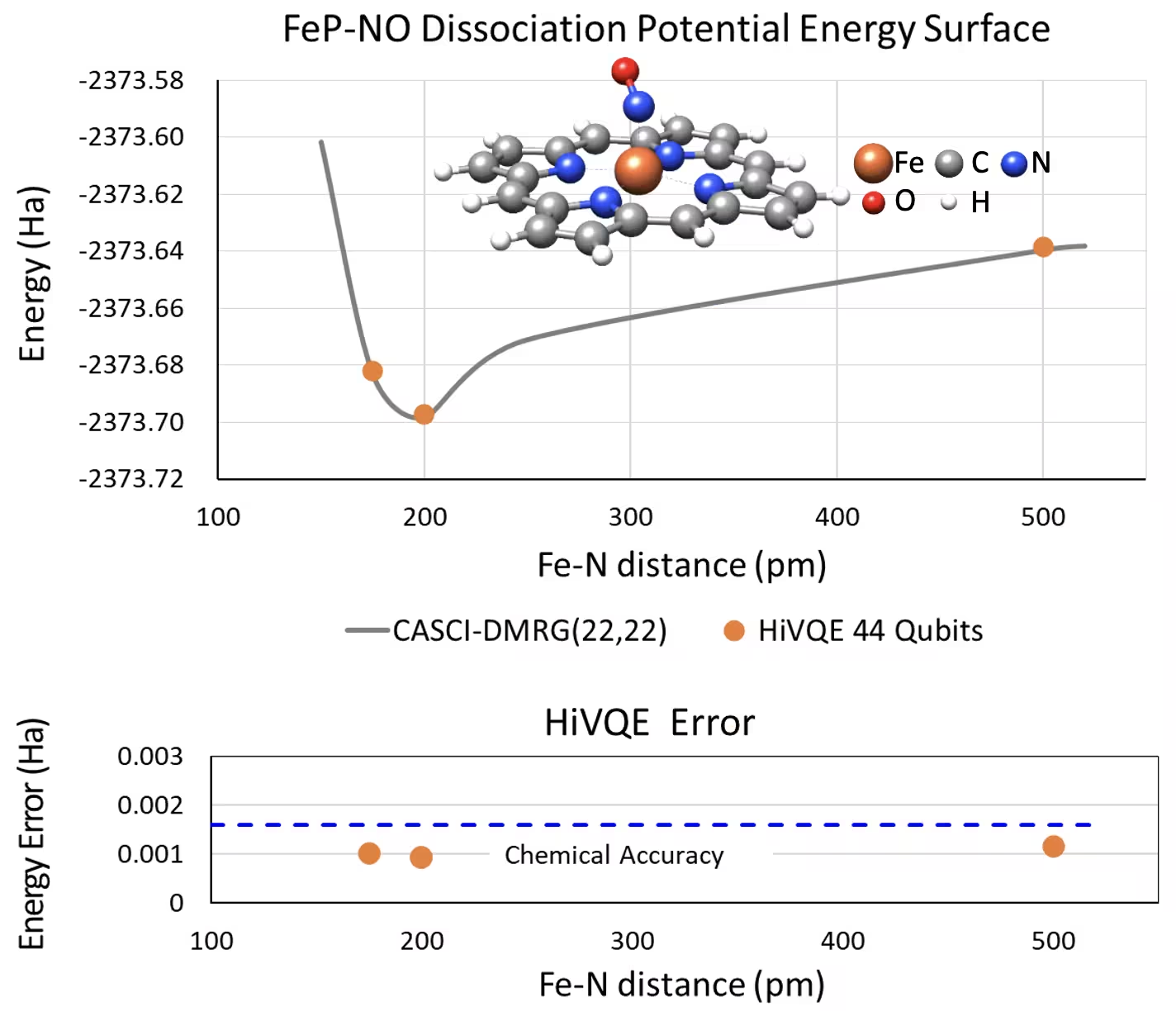
Dissoziations-PES-Kurve für fünffach koordiniertes Eisen(II)-Porphyrin mit einem NO-System mit 44 Qubits
Ein weiteres interessantes chemisches System ist ein Eisen(II)-Porphyrin-Komplex (FeP) mit einem koordinierten Stickstoffmonoxid-Liganden (NO), der ein biologisch relevantes Metalloporphyrinsystem darstellt und in verschiedenen physiologischen Prozessen eine entscheidende Rolle spielt. In diesem Beispiel wurde HI-VQE verwendet, um die genaue Potentialenergieflächenkurve der intermolekularen Wechselwirkung zwischen FeP und NO (Grundzustandsenergie für unterschiedlich separierte Geometrien) zu schätzen. Das kombinierte System hat 450 Orbitale und 202 Elektronen (450o,202e) mit 6-31g(d)-Basis insgesamt. Die homo-lumo aktive Orbitalauswahl wurde verwendet, um den kleineren Fall aus dem realen Fall mit (22o,22e) zu berechnen. Aus den folgenden Benchmark-Ergebnissen konnten wir die chemische Genauigkeit (> 1,6 mHa) mit einer hochmodernen klassischen Computerchemie-Berechnung der CASCI(DMRG) (22o,22e)-Referenz erreichen.
Benchmarks
- Die exakte Matrixgröße ist die Anzahl der Determinanten für die exakte Lösung, z. B. FCI und CASCI.
- HI-VQE sampelt und berechnet den Teilraum davon (d. h. die HI-VQE-Matrixgröße).
- Die Gesamtzeit umfasst die QPU-Laufzeit und Qiskit Function-Läufe mit CPU.
- Die Genauigkeit wird aus der Energiedifferenz zur exakten Lösung geschätzt.
| Chemisches System | Anzahl der Qubits | Exakte Matrixgröße | HI-VQE Matrixgröße | E(diff) zur exakten (mHa) | Anzahl der Iterationen | Gesamtzeit | QPU-Laufzeit |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
Fehlermeldungen abrufen
Wenn deine Arbeitslast fehlschlägt, ist der Status ERROR und der Aufruf von job.result() löst eine Ausnahme aus:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Support erhalten
Du kannst eine E-Mail an qiskit.support@qunovacomputing.com senden, um Hilfe zu dieser Function zu erhalten.
Wenn du Hilfe bei der Fehlerbehebung eines bestimmten Fehlers benötigst, gib bitte die Function Job-ID des Jobs an, bei dem der Fehler aufgetreten ist.
Nächste Schritte
- Beantrage Zugang zur Function, indem du dieses Formular ausfüllst.
- Besuche die API-Referenz für diese Qiskit Function.
- Probiere das Tutorial Compute dissociation PES curve for FeP-NO with HI-VQE aus.
- Lies Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Probiere das Tutorial Dissociation PES curves with Qunova HiVQE aus.