Verbesserung der Energieschätzung eines Chemie-Hamiltonians mit SQD
In diesem Tutorial implementieren wir ein Qiskit-Muster, das zeigt, wie verrauschte Quantensamples nachbearbeitet werden, um eine Näherung des Grundzustands eines Chemie-Hamiltonians zu finden: das -Molekül im Gleichgewicht im 6-31G-Basissatz. Wir folgen einem stichprobenbasierten Quantendiagonalisierungsansatz, um Samples zu verarbeiten, die aus einem 36-Qubit-Quantenschaltkreis-Ansatz (in diesem Fall einem LUCJ-Schaltkreis) entnommen wurden. Um den Effekt von Quantenrauschen zu berücksichtigen, wird die Konfigurationswiederherstellungstechnik verwendet.
Das Muster lässt sich in vier Schritte unterteilen:
- Schritt 1: Auf Quantenproblem abbilden
- Einen Ansatz zur Schätzung des Grundzustands erstellen
- Schritt 2: Das Problem optimieren
- Den Ansatz für das Backend transpilieren
- Schritt 3: Experimente durchführen
- Samples aus dem Ansatz mit dem
Sampler-Primitiv entnehmen
- Samples aus dem Ansatz mit dem
- Schritt 4: Ergebnisse nachbearbeiten
- Selbstkonsistente Konfigurationswiederherstellungsschleife
- Den vollständigen Satz von Bitstring-Samples nachbearbeiten, unter Verwendung von Vorwissen über die Teilchenzahl und die durchschnittliche Orbitalbesetzung, die in der letzten Iteration berechnet wurde.
- Probabilistisch Batches von Teilsamples aus wiederhergestellten Bitstrings erstellen.
- Den molekularen Hamiltonian über jeden abgetasteten Teilraum projizieren und diagonalisieren.
- Die minimale Grundzustandsenergie über alle Batches speichern und die durchschnittliche Orbitalbesetzung aktualisieren.
- Selbstkonsistente Konfigurationswiederherstellungsschleife
Für dieses Beispiel hat der wechselwirkende Elektronen-Hamiltonian die allgemeine Form:
/ sind die fermionischen Erzeugungs-/Vernichtungsoperatoren, die dem -ten Basissatzelement und dem Spin zugeordnet sind. und sind die ein- und zweiteiligen elektronischen Integrale.
Der SQD-Workflow mit selbstkonsistenter Konfigurationswiederherstellung ist im folgenden Diagramm dargestellt.
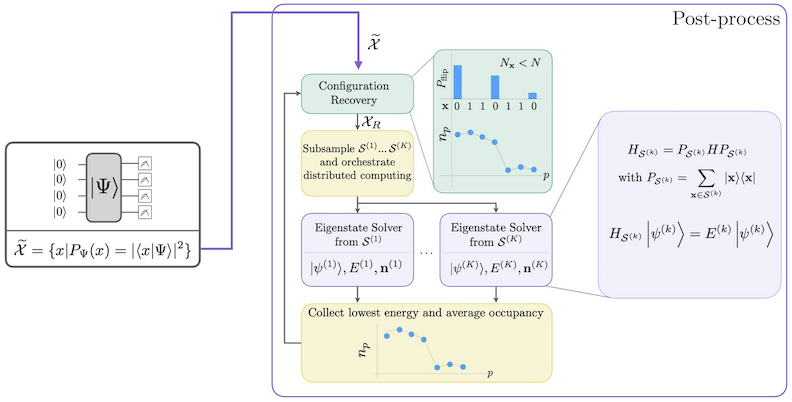
Es ist bekannt, dass SQD gut funktioniert, wenn der Ziel-Eigenzustand dünnbesetzt ist: Die Wellenfunktion ist auf einer Menge von Basiszuständen getragen, deren Größe nicht exponentiell mit der Problemgröße wächst. In diesem Szenario liefert die Diagonalisierung des in den durch definierten Teilraum projizierten Hamiltonians:
eine gute Näherung des Ziel-Eigenzustands. Die Rolle des Quantengeräts besteht darin, Samples der Elemente von zu erzeugen. Zunächst bereitet ein Quantenschaltkreis den Zustand im Quantengerät vor. Die Jordan-Wigner-Kodierung wird verwendet. Folglich repräsentieren Elemente der Rechenbasis Fock-Zustände (elektronische Konfigurationen/Determinanten). Der Schaltkreis wird in der Rechenbasis abgetastet, was den Satz verrauschter Konfigurationen ergibt. Die Konfigurationen werden durch Bitstrings dargestellt. Der Satz wird dann an den klassischen Nachbearbeitungsblock übergeben, wo die selbstkonsistente Konfigurationswiederherstellungstechnik verwendet wird. Im SQD-Framework besteht die Rolle des Quantengeräts darin, eine Wahrscheinlichkeitsverteilung zu erzeugen.
Schritt 1: Problem auf einen Quantenschaltkreis abbilden
In diesem Tutorial werden wir die Grundzustandsenergie eines -Moleküls näherungsweise bestimmen. Zunächst legen wir das Molekül und seine Eigenschaften fest. Als Nächstes erstellen wir einen lokalen unitären Cluster-Jastrow (LUCJ)-Ansatz (Quantenschaltkreis), um Samples von einem Quantencomputer für die Grundzustandsenergieschätzung zu erzeugen.
Zuerst legen wir das Molekül und seine Eigenschaften fest.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Als Nächstes erstellen wir den Ansatz. Der LUCJ-Ansatz ist ein parametrisierter Quantenschaltkreis, und wir initialisieren ihn mit t2- und t1-Amplituden, die aus einer CCSD-Berechnung gewonnen wurden.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Wir werden das ffsim-Paket verwenden, um den Ansatz zu erstellen und ihn mit den oben berechneten t2- und t1-Amplituden zu initialisieren. Da unser Molekül einen geschlossenschaligen Hartree-Fock-Zustand hat, verwenden wir die spinausgeglichene Variante des UCJ-Ansatzes, UCJOpSpinBalanced.
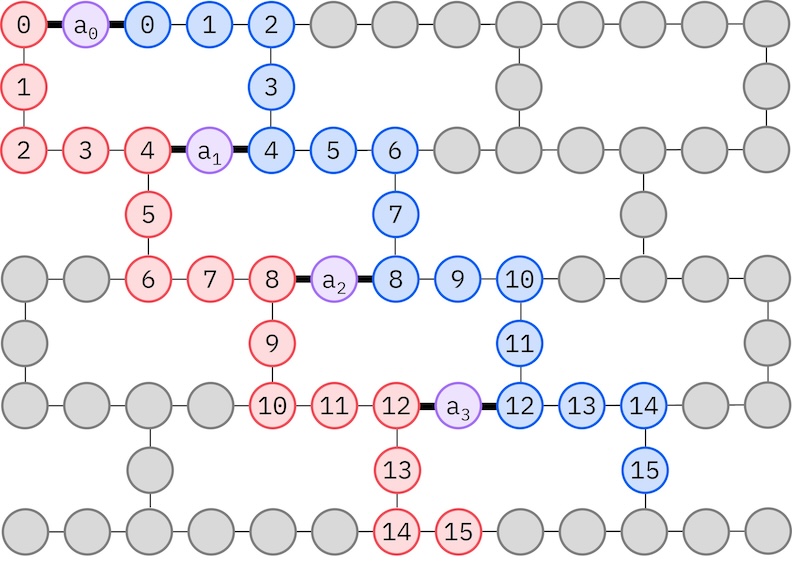
Da unsere Ziel-IBM-Hardware eine Heavy-Hex-Topologie hat, übernehmen wir das Zickzack-Muster für Qubit-Wechselwirkungen. In diesem Muster sind Orbitale (dargestellt durch Qubits) mit demselben Spin mit einer Linientopologie verbunden (rote und blaue Kreise), wobei jede Linie aufgrund der Heavy-Hex-Konnektivität der Zielhardware eine Zickzackform annimmt. Ebenfalls aufgrund der Heavy-Hex-Topologie haben Orbitale verschiedener Spins Verbindungen zwischen jedem 4. Orbital (0, 4, 8, usw.) (lila Kreise).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Schritt 2: Das Problem optimieren
Als Nächstes optimieren wir unseren Schaltkreis für eine Zielhardware. Wir müssen das zu verwendende Hardware-Gerät auswählen, bevor wir unseren Schaltkreis optimieren. Wir verwenden ein simuliertes 127-Qubit-Backend aus qiskit_ibm_runtime, um ein echtes Gerät zu emulieren. Um auf einer echten QPU zu laufen, ersetze einfach das simulierte Backend durch ein echtes Backend. Weitere Informationen findest du in den Qiskit IBM Runtime-Dokumentationen.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Als Nächstes empfehlen wir die folgenden Schritte, um den Ansatz zu optimieren und hardwarekompatibel zu machen.
- Physikalische Qubits (
initial_layout) von der Zielhardware auswählen, die dem oben beschriebenen Zickzack-Muster entsprechen. Die Anordnung von Qubits in diesem Muster führt zu einem effizienten, hardwarekompatiblen Schaltkreis mit weniger Gates. - Einen gestuften Pass-Manager mit der Funktion generate_preset_pass_manager aus Qiskit mit deiner Wahl von
backendundinitial_layoutgenerieren. - Die
pre_init-Stufe deines gestuften Pass-Managers aufffsim.qiskit.PRE_INITsetzen.ffsim.qiskit.PRE_INITenthält Qiskit-Transpiler-Passes, die Gates in Orbitalrotationen zerlegen und diese dann zusammenführen, was zu weniger Gates im endgültigen Schaltkreis führt. - Den Pass-Manager auf deinen Schaltkreis anwenden.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Schritt 3: Experimente durchführen
Nach der Optimierung des Schaltkreises für die Hardware-Ausführung sind wir bereit, ihn auf der Zielhardware auszuführen und Samples für die Grundzustandsenergieschätzung zu sammeln. Da wir nur einen Schaltkreis haben, verwenden wir den Job-Ausführungsmodus von Qiskit Runtime und führen unseren Schaltkreis aus.
Hinweis: Wir haben den Code zur Ausführung des Schaltkreises auf einer QPU auskommentiert und ihn als Referenz für den Benutzer belassen. Anstatt in diesem Leitfaden auf echter Hardware zu laufen, werden wir einfach zufällige Samples erzeugen, die aus der Gleichverteilung gezogen wurden.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Schritt 4: Ergebnisse nachbearbeiten
Jetzt führen wir den SQD-Algorithmus mit der Funktion diagonalize_fermionic_hamiltonian aus. Erläuterungen zu den Argumenten dieser Funktion findest du in der API-Dokumentation.
Der im SQD-Addon enthaltene Solver verwendet die PySCF-Implementierung von Selected CI, insbesondere pyscf.fci.selected_ci.kernel_fixed_space. Das folgende Beispiel zeigt auch, wie Schlüsselwortargumente über den enthaltenen Solver an diese Funktion übergeben werden. Hier übergeben wir das Argument max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
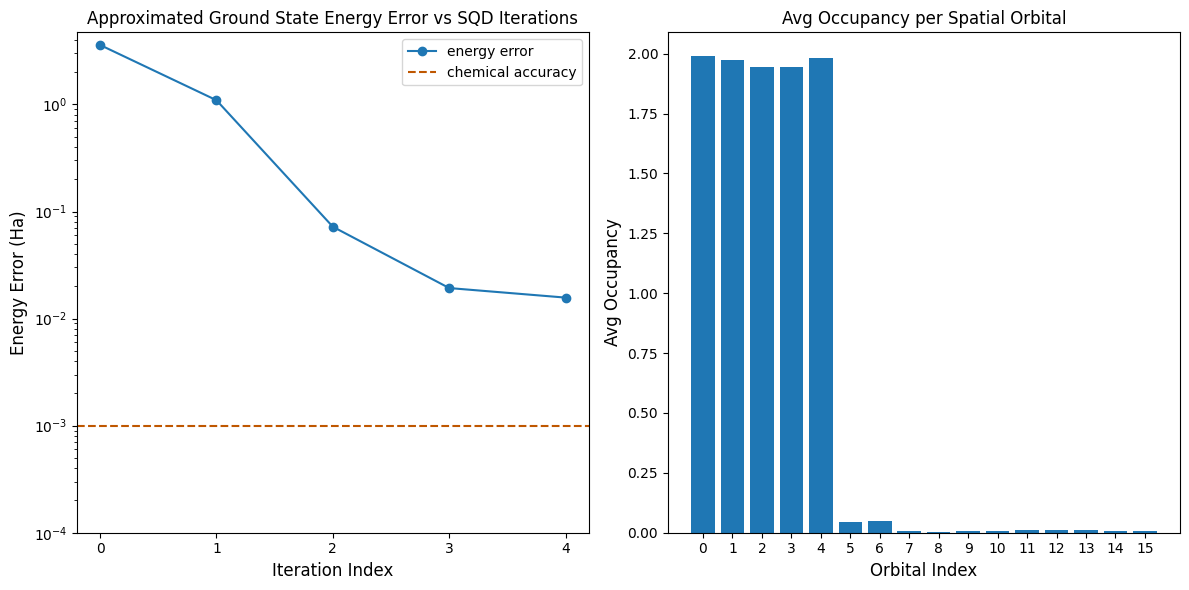
Jetzt stellen wir die Ergebnisse grafisch dar.
Der erste Plot zeigt, dass wir nach wenigen Iterationen die Grundzustandsenergie auf ~16 mH genau schätzen (chemische Genauigkeit wird typischerweise als 1 kcal/mol 1.6 mH akzeptiert). Bedenke, dass die Quantensamples in dieser Demo reines Rauschen waren. Das Signal hier stammt aus dem a-priori-Wissen über die elektronische Struktur und den molekularen Hamiltonian.
Der zweite Plot zeigt die durchschnittliche Besetzung jedes räumlichen Orbitals nach der letzten Iteration. Wir können sehen, dass sowohl die spin-up- als auch die spin-down-Elektronen mit hoher Wahrscheinlichkeit die ersten fünf Orbitale in unseren Lösungen besetzen.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha