Der Variational Quantum Eigensolver (VQE)
Diese Lektion stellt den Variational Quantum Eigensolver vor, erläutert seine Bedeutung als grundlegenden Algorithmus im Quantencomputing und untersucht seine Stärken und Schwächen. VQE allein, ohne ergänzende Methoden, wird für moderne Quantenberechnungen auf Utility-Skala wahrscheinlich nicht ausreichen. Er ist dennoch wichtig als archetypische klassisch-quantenmechanische Hybridmethode und bildet ein wesentliches Fundament, auf dem viele fortgeschrittenere Algorithmen aufbauen.
Dieses Video gibt einen Überblick über VQE und die Faktoren, die seine Effizienz beeinflussen. Der folgende Text ergänzt dies um weitere Details und implementiert VQE mit Qiskit.
1. Was ist VQE?
Der Variational Quantum Eigensolver ist ein Algorithmus, der klassisches und quantenmechanisches Computing gemeinsam einsetzt, um eine Aufgabe zu lösen. Eine VQE-Berechnung besteht aus vier Hauptkomponenten:
- Ein Operator: Oft ein Hamiltonoperator, den wir nennen, der eine Eigenschaft des Systems beschreibt, die du optimieren möchtest. Anders ausgedrückt: Du suchst den Eigenvektor dieses Operators, der dem minimalen Eigenwert entspricht. Diesen Eigenvektor nennen wir oft den „Grundzustand".
- Ein „Ansatz" (ein deutsches Wort): Ein Quanten-Circuit, der einen Quantenzustand vorbereitet, der den gesuchten Eigenvektor annähert. Genau genommen ist ein Ansatz eine Familie von Quanten-Circuits, weil einige der Gates im Ansatz parametrisiert sind – ihnen wird also ein Parameter übergeben, den wir variieren können. Diese Familie von Circuits kann eine Familie von Quantenzuständen erzeugen, die den Grundzustand approximieren.
- Ein Estimator: Ein Mittel zur Schätzung des Erwartungswerts des Operators über den aktuellen variationellen Quantenzustand. Manchmal ist dieser Erwartungswert – die sogenannte Kostenfunktion – unser eigentliches Ziel. Manchmal interessiert uns eine komplexere Funktion, die sich dennoch aus einem oder mehreren Erwartungswerten zusammensetzt.
- Ein klassischer Optimierer: Ein Algorithmus, der Parameter variiert, um die Kostenfunktion zu minimieren.
Schauen wir uns jede dieser Komponenten genauer an.
1.1 Der Operator (Hamiltonoperator)
Im Mittelpunkt eines VQE-Problems steht ein Operator, der ein System von Interesse beschreibt. Wir nehmen hier an, dass der kleinste Eigenwert und der zugehörige Eigenvektor dieses Operators für einen wissenschaftlichen oder praktischen Zweck nützlich sind. Beispiele hierfür sind ein chemischer Hamiltonoperator, der ein Molekül beschreibt, bei dem der kleinste Eigenwert der Grundzustandsenergie des Moleküls entspricht und der zugehörige Eigenzustand die Geometrie oder Elektronenkonfiguration des Moleküls beschreibt. Oder der Operator beschreibt die Kosten eines zu optimierenden Prozesses, und die Eigenzustände entsprechen Routen oder Vorgehensweisen. In manchen Fachgebieten wie der Physik bezeichnet ein „Hamiltonoperator" fast immer einen Operator, der die Energie eines physikalischen Systems beschreibt. Im Quantencomputing ist es jedoch üblich, quantenmechanische Operatoren, die ein betriebswirtschaftliches oder logistisches Problem beschreiben, ebenfalls als „Hamiltonoperator" zu bezeichnen. Diese Konvention übernehmen wir hier.

Die Abbildung eines physikalischen oder Optimierungsproblems auf Qubits ist typischerweise keine triviale Aufgabe, aber diese Details stehen nicht im Mittelpunkt dieses Kurses. Eine allgemeine Diskussion der Abbildung eines Problems auf einen Quantenoperator findet sich in Quantum computing in practice. Ein detaillierterer Blick auf die Abbildung chemischer Probleme auf Quantenoperatoren bietet Quantum Chemistry with VQE.
Für die Zwecke dieses Kurses nehmen wir an, dass die Form des Hamiltonoperators bekannt ist. Ein Hamiltonoperator für ein einfaches Wasserstoffmolekül (unter bestimmten Aktivraum-Annahmen und mit dem Jordan-Wigner-Mapper) lautet beispielsweise:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Beachte, dass der obige Hamiltonoperator Terme wie ZZII und YYYY enthält, die nicht miteinander kommutieren. Um ZZII auszuwerten, müsste man unter anderem den Pauli-Z-Operator auf Qubit 3 messen. Um jedoch YYYY auszuwerten, muss man den Pauli-Y-Operator auf demselben Qubit 3 messen. Zwischen dem Y- und dem Z-Operator auf demselben Qubit besteht eine Unschärferelation; beide Operatoren lassen sich nicht gleichzeitig messen. Auf diesen Punkt werden wir weiter unten und im weiteren Verlauf des Kurses noch zurückkommen.
Der obige Hamiltonoperator ist eine -Matrixoperator. Die Diagonalisierung des Operators zur Bestimmung seines kleinsten Energieeigenwerts ist nicht schwierig.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
Klassische Brute-Force-Eigenlöser können nicht skalieren, um die Energien oder Geometrien sehr großer Atomsysteme wie Medikamente oder Proteine zu beschreiben. VQE ist einer der frühen Versuche, Quantencomputing für dieses Problem nutzbar zu machen.
In dieser Lektion werden wir auf deutlich größere Hamiltonoperatoren stoßen als den obigen. Es wäre jedoch verfrüht, die Grenzen von VQE auszuloten, bevor wir später in diesem Kurs einige der fortgeschritteneren Werkzeuge vorstellen, die VQE ergänzen oder ersetzen können.
1.2 Ansatz
Das Wort „Ansatz" ist deutsch. Der korrekte Plural im Deutschen lautet „Ansätze", obwohl man häufig auch „ansatzes" oder „ansatze" findet. Im Kontext von VQE ist ein Ansatz der Quanten-Circuit, mit dem du eine Mehr-Qubit-Wellenfunktion erzeugst, die den Grundzustand des untersuchten Systems möglichst gut annähert und damit den kleinsten Erwartungswert deines Operators liefert. Dieser Circuit enthält variationelle Parameter (oft im Parametervektor zusammengefasst).

Es wird ein Anfangssatz von Werten der variationellen Parameter gewählt. Die unitäre Operation des Ansatzes auf dem Circuit nennen wir . Standardmäßig werden alle Qubits in IBM®-Quantencomputern im Zustand initialisiert. Wenn der Circuit ausgeführt wird, ist der Zustand der Qubits:
Wenn wir nur die niedrigste Energie benötigten (in der Sprache physikalischer Systeme), könnten wir diese schätzen, indem wir die Energie viele Male messen und den kleinsten Wert nehmen. Wir wollen aber typischerweise auch die Konfiguration kennen, die diese niedrigste Energie oder diesen kleinsten Eigenwert liefert. Der nächste Schritt ist daher die Schätzung des Erwartungswerts des Hamiltonoperators durch Quantenmessungen. Darin steckt einiges. Qualitativ lässt sich dieser Prozess damit verstehen, dass die Wahrscheinlichkeit , eine Energie zu messen (wieder in der Sprache physikalischer Systeme), mit dem Erwartungswert durch folgende Beziehung verknüpft ist:
Die Wahrscheinlichkeit ist außerdem mit der Überlappung zwischen dem Eigenzustand und dem aktuellen Systemzustand verknüpft:
Durch viele Messungen der Pauli-Operatoren, aus denen unser Hamiltonoperator besteht, können wir den Erwartungswert des Hamiltonoperators im aktuellen Systemzustand schätzen. Der nächste Schritt besteht darin, die Parameter zu variieren und sich dem Grundzustand des Systems mit der niedrigsten Energie noch weiter anzunähern. Wegen der variationellen Parameter im Ansatz wird er manchmal auch als Variationsform bezeichnet.
Bevor wir uns diesem Variationsprozess zuwenden, ist es oft nützlich, von einem „guten Ausgangszustand" zu starten. Wenn du genug über dein System weißt, kannst du eine bessere Anfangsschätzung als machen. Zum Beispiel ist es in chemischen Anwendungen üblich, Qubits im Hartree-Fock-Zustand zu initialisieren. Dieser Ausgangszustand ohne variationelle Parameter heißt Referenzzustand. Den Circuit zur Erzeugung des Referenzzustands nennen wir . Wann immer es wichtig ist, den Referenzzustand vom restlichen Ansatz zu unterscheiden, verwende: Äquivalent gilt:
1.3 Estimator
Wir brauchen eine Möglichkeit, den Erwartungswert unseres Hamiltonoperators in einem bestimmten variationellen Zustand zu schätzen. Könnten wir den gesamten Operator direkt messen, wäre dies so einfach wie viele (sagen wir ) Messungen durchzuführen und die gemessenen Werte zu mitteln:
Das -Symbol erinnert uns daran, dass dieser Erwartungswert nur im Grenzfall exakt wäre. Mit tausenden von Messungen an einem Circuit ist der Stichprobenfehler des Erwartungswerts jedoch recht gering. Für sehr präzise Berechnungen spielen andere Faktoren wie Rauschen eine Rolle.
Es ist jedoch im Allgemeinen nicht möglich, auf einmal zu messen. kann mehrere nicht-kommutierende Pauli-X-, Y- und Z-Operatoren enthalten. Daher muss der Hamiltonoperator in Gruppen von Operatoren aufgeteilt werden, die gleichzeitig gemessen werden können, und jede solche Gruppe muss separat geschätzt werden; anschließend werden die Ergebnisse kombiniert, um einen Erwartungswert zu erhalten. Wir werden dies in der nächsten Lektion, wenn wir die Skalierung klassischer und quantenmechanischer Ansätze besprechen, ausführlicher behandeln. Diese Komplexität bei der Messung ist ein Grund, warum wir hocheffizienten Code für diese Art von Schätzung benötigen. In dieser und den folgenden Lektionen verwenden wir dafür das Qiskit-Runtime-Primitiv Estimator.
1.4 Klassische Optimierer
Ein klassischer Optimierer ist ein klassischer Algorithmus, der darauf ausgelegt ist, Extremwerte einer Zielfunktion zu finden (typischerweise ein Minimum). Er durchsucht den Raum möglicher Parameter nach einem Satz, der eine interessierende Funktion minimiert. Grob lassen sich solche Algorithmen in gradientenbasierte Methoden, die Gradienteninformationen nutzen, und gradientenfreie Methoden einteilen, die als Black-Box-Optimierer arbeiten. Die Wahl des klassischen Optimierers kann die Leistung eines Algorithmus erheblich beeinflussen, insbesondere in Gegenwart von Rauschen in Quantenhardware. Zu den gängigen Optimierern in diesem Bereich gehören Adam, AMSGrad und SPSA, die in verrauschten Umgebungen vielversprechende Ergebnisse gezeigt haben. Zu den traditionelleren Optimierern zählen COBYLA und SLSQP.
Ein typischer Workflow (wie in Abschnitt 3.3 gezeigt) besteht darin, einen dieser Algorithmen als Methode innerhalb eines Minimierers wie SciPys minimize-Funktion zu verwenden. Diese nimmt als Argumente entgegen:
- Eine zu minimierende Funktion. Oft ist das der Erwartungswert der Energie. Im Allgemeinen werden diese als „Kostenfunktionen" bezeichnet.
- Einen Satz von Startparametern für die Suche. Häufig als oder bezeichnet.
- Argumente, einschließlich der Argumente der Kostenfunktion. Im Quantencomputing mit Qiskit umfassen diese den Ansatz, den Hamiltonoperator und das Estimator-Primitiv, das im nächsten Unterabschnitt näher beschrieben wird.
- Eine Minimierungsmethode (
method). Dies ist der spezifische Algorithmus, der den Parameterraum durchsucht. Hier würde man zum Beispiel COBYLA oder SLSQP angeben. - Optionen (
options). Die verfügbaren Optionen können je nach Methode unterschiedlich sein. Ein Beispiel, das praktisch alle Methoden beinhalten, ist die maximale Anzahl von Iterationen des Optimierers vor dem Abbruch:maxiter.

Bei jedem iterativen Schritt wird der Erwartungswert des Hamiltonoperators durch viele Messungen geschätzt. Dieser geschätzte Energiewert wird von der Kostenfunktion zurückgegeben, und der Minimierer aktualisiert seine Information über die Energielandschaft. Wie genau der Optimierer den nächsten Schritt wählt, hängt von der Methode ab. Manche nutzen Gradienten und wählen die Richtung des steilsten Abstiegs. Andere berücksichtigen Rauschen und verlangen, dass die Kosten um eine große Marge sinken, bevor sie akzeptieren, dass die wahre Energie in dieser Richtung abnimmt.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 Das Variationsprinzip
Das Variationsprinzip ist in diesem Kontext von zentraler Bedeutung: Es besagt, dass keine variationelle Wellenfunktion einen Energie- (oder Kosten-)Erwartungswert liefern kann, der kleiner ist als der der Grundzustandswellenfunktion. Mathematisch ausgedrückt:
Dies lässt sich leicht nachweisen, wenn man beachtet, dass die Menge aller Eigenzustände von eine vollständige Basis für den Hilbertraum bilden. Mit anderen Worten: Jeder Zustand, insbesondere , lässt sich als gewichtete (normierte) Summe dieser Eigenzustände von schreiben:
wobei zu bestimmende Konstanten sind und gilt. Wir überlassen dies als Übungsaufgabe. Beachte jedoch die Konsequenz: Der variationelle Zustand, der den kleinsten Erwartungswert der Energie liefert, ist die beste Näherung des wahren Grundzustands.
Verständnisfragen
Zeige mathematisch, dass für jeden variationellen Zustand gilt.
Antwort
Mit der gegebenen Entwicklung des variationellen Zustands in Energie-Eigenzustände,
können wir den variationellen Energieerwartungswert schreiben als
Für alle Koeffizienten gilt . Damit ergibt sich:
2. Vergleich mit dem klassischen Workflow
Angenommen, du interessierst dich für eine Matrix mit N Zeilen und N Spalten. Stell dir vor, deine Matrix ist so groß, dass eine exakte Diagonalisierung keine Option ist. Angenommen, du weißt genug über dein Problem, um einige Vermutungen über die allgemeine Struktur des Ziel-Eigenzustands anzustellen, und du möchtest Zustände ähnlich deiner Anfangsschätzung untersuchen, um zu sehen, ob deine Kosten/Energie weiter gesenkt werden können. Dies ist ein variationeller Ansatz und eine Methode, die eingesetzt wird, wenn eine exakte Diagonalisierung keine Option ist.
2.1 Klassischer Workflow
Mit einem klassischen Computer würde dies folgendermaßen funktionieren:
- Mache einen Schätzzustand mit einigen Parametern , die du variieren wirst: . Obwohl diese Anfangsschätzung zufällig sein könnte, ist das nicht ratsam. Wir wollen das Wissen über das vorliegende Problem nutzen, um unsere Schätzung so weit wie möglich anzupassen.
- Berechne den Erwartungswert des Operators, wenn sich das System in diesem Zustand befindet:
- Ändere die variationellen Parameter und wiederhole: .
- Nutze die gesammelten Informationen über die Landschaft möglicher Zustände in deinem variationellen Teilraum, um immer bessere Schätzungen zu machen und dich dem Zielzustand anzunähern. Das Variationsprinzip garantiert, dass unser variationeller Zustand keinen Eigenwert liefern kann, der kleiner ist als der des angestrebten Grundzustands. Je kleiner also der Erwartungswert, desto besser unsere Näherung des Grundzustands:
Betrachten wir die Schwierigkeit jedes Schritts in diesem Ansatz. Das Setzen oder Aktualisieren von Parametern ist rechnerisch einfach; die Schwierigkeit liegt darin, nützliche, physikalisch motivierte Anfangsparameter zu wählen. Die gesammelten Informationen aus früheren Iterationen zu nutzen, um Parameter so zu aktualisieren, dass man sich dem Grundzustand nähert, ist nicht trivial. Es gibt jedoch klassische Optimierungsalgorithmen, die dies recht effizient tun. Diese klassische Optimierung ist nur deshalb aufwändig, weil sie viele Iterationen erfordern kann; im schlimmsten Fall kann die Anzahl der Iterationen exponentiell mit N skalieren. Der rechenintensivste einzelne Schritt ist mit großer Wahrscheinlichkeit die Berechnung des Erwartungswerts deiner Matrix mit einem gegebenen Zustand :
Die -Matrix muss auf den -elementigen Vektor wirken, was im schlimmsten Fall Multiplikationsoperationen entspricht. Dies muss bei jeder Parameteriteration durchgeführt werden. Für extrem große Matrizen ist dies mit hohen Rechenkosten verbunden.
2.2 Quantenworkflow und kommutierende Pauli-Gruppen
Stell dir nun vor, du überträgst diesen Teil der Berechnung an einen Quantencomputer. Anstatt diesen Erwartungswert zu berechnen, schätzt du ihn, indem du den Zustand auf dem Quantencomputer mit deinem variationellen Ansatz vorbereitest und dann Messungen durchführst.
Das klingt einfacher, als es ist. ist im Allgemeinen nicht leicht zu messen. Er könnte beispielsweise aus vielen nicht-kommutierenden Pauli-X-, Y- und Z-Operatoren bestehen. Aber kann als Linearkombination von Termen geschrieben werden, von denen jeder leicht messbar ist (zum Beispiel Pauli-Operatoren oder Gruppen von qubitweise kommutierenden Pauli-Operatoren). Der Erwartungswert von über einen Zustand ist die gewichtete Summe der Erwartungswerte der konstituierenden Terme . Dieser Ausdruck gilt für jeden Zustand , wir werden ihn aber speziell mit unseren variationellen Zuständen verwenden.
wobei ein Pauli-String wie IZZX…XIYX ist, oder mehrere solcher Strings, die miteinander kommutieren. Eine Beschreibung des Erwartungswerts, die der Realität von Messungen auf Quantencomputern näher kommt, lautet daher:
Und im Kontext unserer variationellen Wellenfunktion:
Jeder der Terme kann Mal gemessen werden und liefert Messstichproben mit sowie einen Erwartungswert und eine Standardabweichung . Diese Terme können summiert und die Fehler durch die Summe propagiert werden, um einen Gesamterwartungswert und eine Gesamtstandardabweichung zu erhalten.
Dies erfordert keine aufwändigen Matrixmultiplikationen und auch keinen Prozess, der notwendigerweise wie skaliert. Stattdessen sind mehrere Messungen auf dem Quantencomputer erforderlich. Wenn nicht zu viele davon benötigt werden, könnte dieser Ansatz effizient sein. Das ist der quantenmechanische Teil von VQE.
Sprechen wir aber über Gründe, warum dies möglicherweise nicht effizient ist. Ein Grund für viele Messungen ist die Reduzierung der statistischen Unsicherheit bei Schätzungen, die für sehr präzise Berechnungen erforderlich ist. Ein weiterer Grund ist die Anzahl der Pauli-Strings, die benötigt werden, um die gesamte Matrix abzudecken. Da Pauli-Matrizen (plus die Identität: X, Y, Z und I) den Raum aller Operatoren einer bestimmten Dimension aufspannen, ist garantiert, dass wir unsere interessierende Matrix als gewichtete Summe von Pauli-Operatoren schreiben können, wie wir es zuvor getan haben.
wobei ein Pauli-String ist, der auf alle Qubits des Systems wirkt, wie IZZX…XIYX, oder mehrere solcher Strings, die miteinander kommutieren. Beachte, dass Qiskit die Little-Endian-Notation verwendet, bei der der Pauli-Operator von rechts auf das Qubit wirkt. Wir können unseren Operator also messen, indem wir eine Reihe von Pauli-Operatoren messen.
Wir können diese Pauli-Operatoren jedoch nicht alle gleichzeitig messen. Pauli-Operatoren (ohne I) kommutieren nicht miteinander, wenn sie mit demselben Qubit verknüpft sind. Zum Beispiel können wir IZIZ und ZZXZ gleichzeitig messen, weil wir I und Z gleichzeitig für das dritte Qubit messen können, und I und X gleichzeitig für das erste Qubit. Aber wir können ZZZZ und ZZZX nicht gleichzeitig messen, weil Z und X nicht kommutieren und beide auf das 0. Qubit wirken. Erfahrene Leserinnen und Leser erinnern sich vielleicht daran, dass zwei Gruppen von Pauli-Operatoren als Menge kommutieren können, auch wenn die Messungen der einzelnen Qubits nicht kommutieren. Der Estimator geht von Tensor-Produkt-Pauli-Messungen aus (über Basisrotationen), was der Gruppierung qubit-weise kommutierender Operatoren entspricht. Um also zwei Strings (A und B) von Pauli-Operatoren mit dem Estimator gleichzeitig zu schätzen, müssen die Pauli-Operatoren jedes Qubits in A und B kommutieren. Das bedeutet, dass wir auch ZZZZ und ZZXX nicht gleichzeitig messen können.

Wir zerlegen also unsere Matrix in eine Summe von Paulis, die auf verschiedene Qubits wirken. Einige Elemente dieser Summe können gleichzeitig gemessen werden; wir nennen dies eine Gruppe kommutierender Paulis. Je nachdem, wie viele nicht-kommutierende Terme es gibt, benötigen wir möglicherweise viele solcher Gruppen. Die Anzahl solcher Gruppen kommutierender Pauli-Strings nennen wir . Wenn klein ist, könnte dies gut funktionieren. Hat Millionen von Gruppen, wird dies nicht nützlich sein.
Die für die Schätzung des Erwartungswerts erforderlichen Prozesse sind im Qiskit-Runtime-Primitiv Estimator zusammengefasst. Mehr über den Estimator erfährst du in der API-Referenz in der IBM Quantum®-Dokumentation. Man kann den Estimator zwar direkt verwenden, aber er gibt mehr zurück als nur den kleinsten Energieeigenwert – zum Beispiel auch Informationen über den Standard-Stichprobenfehler. Im Kontext von Minimierungsproblemen sieht man den Estimator daher oft innerhalb einer Kostenfunktion. Mehr über Estimator-Eingaben und -Ausgaben erfährst du in diesem Leitfaden in der IBM Quantum-Dokumentation.
Du zeichnest den Erwartungswert (oder die Kostenfunktion) für den Parametersatz , der in deinem Zustand verwendet wird, auf und aktualisierst dann die Parameter. Im Laufe der Zeit könntest du die geschätzten Erwartungswerte oder Kostenfunktionswerte nutzen, um einen Gradienten deiner Kostenfunktion im Teilraum der durch deinen Ansatz abgetasteten Zustände zu approximieren. Es gibt sowohl gradientenbasierte als auch gradientenfreie klassische Optimierer. Beide können unter Trainierbarkeitsroblemen leiden, wie mehreren lokalen Minima und großen Bereichen des Parameterraums mit nahezu null Gradient, die als Barren Plateaus bezeichnet werden.

2.3 Faktoren, die die Rechenkosten bestimmen
VQE wird nicht alle deine schwierigsten Quantenchemieprobleme lösen. Das stimmt. Aber besser bei allen Berechnungen zu sein ist auch nicht der Punkt. Wir haben verändert, was die Rechenkosten bestimmt.

Wir haben uns von einem Prozess, dessen Komplexität nur von der Matrixdimension abhängt, zu einem Prozess verlagert, der von der erforderlichen Präzision und der Anzahl nicht-kommutierender Pauli-Operatoren abhängt, aus denen die Matrix besteht. Der letzte Aspekt hat kein Analogon im klassischen Computing.
Basierend auf diesen Abhängigkeiten könnte dieser Prozess für dünnbesetzte Matrizen oder Matrizen mit wenigen nicht-kommutierenden Pauli-Strings nützlich sein. Dies ist beispielsweise bei Systemen wechselwirkender Spins der Fall. Für dichte Matrizen ist er möglicherweise weniger nützlich. Wir wissen zum Beispiel, dass chemische Systeme oft Hamiltonoperatoren haben, die Hunderte, Tausende oder sogar Millionen von Pauli-Strings umfassen. Es gibt interessante Arbeiten, um diese Anzahl von Termen zu reduzieren. Aber chemische Systeme könnten besser zu einigen der anderen Algorithmen passen, die wir in diesem Kurs besprechen werden.
Verständnisfragen
Betrachte einen Hamiltonoperator auf vier Qubits, der folgende Terme enthält:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Du möchtest diese Terme in Gruppen sortieren, sodass alle Terme einer Gruppe gleichzeitig gemessen werden können. Was ist die kleinste Anzahl solcher Gruppen, die du bilden kannst, sodass alle Terme berücksichtigt sind?
Antwort
Es ist in 4 Gruppen möglich. Beachte, dass solche Lösungen typischerweise nicht eindeutig sind.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
Was erwartest du, macht Quantenchemie mit VQE typischerweise schwierig: die Anzahl der Terme im Hamiltonoperator oder das Finden eines guten Ansatzes?
Antwort
Es gibt Ansätze, die für chemische Kontexte hochoptimiert sind. Die Anzahl der Terme im Hamiltonoperator und damit die Anzahl der erforderlichen Messungen verursachen typischerweise mehr Probleme.
3. Beispiel-Hamiltonoperator
Setzen wir diesen Algorithmus anhand eines kleinen Hamiltonoperators in die Praxis um, damit wir sehen können, was in jedem Schritt passiert. Wir verwenden das Qiskit-Patterns-Framework:
-Schritt 1: Problem auf Quanten-Circuits und Operatoren abbilden -Schritt 2: Für Zielhardware optimieren -Schritt 3: Auf Zielhardware ausführen -Schritt 4: Ergebnisse nachverarbeiten
3.1 Schritt 1: Das Problem auf Quantenschaltkreise und Operatoren abbilden
Wir verwenden den oben aus dem Chemiekontext definierten Hamiltonoperator. Zunächst einige allgemeine Imports.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Wir gehen davon aus, dass der gesuchte Hamiltonoperator bekannt ist. Hier verwenden wir einen sehr kleinen Hamiltonoperator, da andere in diesem Kurs besprochene Methoden größere Probleme effizienter lösen können.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
In Qiskit gibt es viele vorgefertigte Ansatz-Optionen. Wir verwenden efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
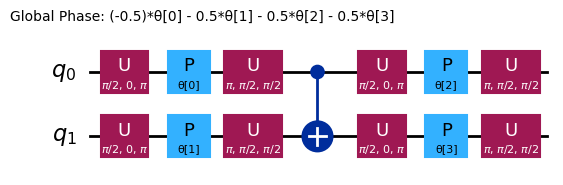
Verschiedene Ansätze haben unterschiedliche Verschränkungsstrukturen und unterschiedliche Rotationsgatter. Der hier gezeigte verwendet CNOT-Gatter zur Verschränkung sowie Y-Gatter und parametrisierte RZ-Gatter für Rotationen. Beachte die Größe dieses Parameterraums: Wir müssen die Kostenfunktion über 4 Variablen (die Parameter der RZ-Gatter) minimieren. Dies lässt sich skalieren, jedoch nicht unbegrenzt. Ein ähnliches Problem auf 4 Qubits mit den Standard-3-Wiederholungen von efficient_su2 ergibt 16 Variationsparameter.
3.2 Schritt 2: Für die Zielhardware optimieren
Der Ansatz wurde mit bekannten Gattern geschrieben, aber unser Circuit muss transpiliert werden, um die Basisgatter nutzen zu können, die auf dem jeweiligen Quantencomputer implementiert sind. Wir wählen das am wenigsten ausgelastete Backend.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Wir können unseren Circuit nun für diese Hardware transpilieren und den transpilierten Ansatz visualisieren.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Beachte, dass sich die verwendeten Gatter geändert haben und die Qubits unseres abstrakten Circuits auf anders nummerierte Qubits des Quantencomputers abgebildet wurden. Um sinnvolle Ergebnisse zu erhalten, müssen wir unseren Hamiltonoperator auf dieselbe Weise abbilden.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Schritt 3: Auf der Zielhardware ausführen
3.3.1 Werte ausgeben
Wir definieren hier eine Kostenfunktion, die als Argumente die in den vorherigen Schritten erstellten Strukturen entgegennimmt: die Parameter, den Ansatz und den Hamiltonoperator. Sie verwendet außerdem Estimator, den wir noch nicht definiert haben. Um das Konvergenzverhalten zu überprüfen, fügen wir Code hinzu, der den Verlauf der Kostenfunktion aufzeichnet.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
Es ist sehr vorteilhaft, wenn du die anfänglichen Parameterwerte auf Basis von Kenntnissen über das jeweilige Problem und die Eigenschaften des Zielzustands wählen kannst. Wir treffen keine solchen Annahmen und verwenden zufällige Anfangswerte.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Wir können uns die Rohausgaben ansehen.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Schritt 4: Ergebnisse nachverarbeiten
Wenn das Verfahren korrekt terminiert, sollten die Werte in unserem Dictionary dem Lösungsvektor bzw. der Gesamtanzahl der Funktionsauswertungen entsprechen. Das lässt sich leicht überprüfen:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum bietet weitere Lernressourcen rund um VQE. Wenn du VQE in die Praxis umsetzen möchtest, sieh dir unser Tutorial an: Grundzustandsenergie-Schätzung der Heisenberg-Kette mit VQE. Weitere Informationen zur Erstellung molekularer Hamiltonoperatoren findest du in dieser Lektion unseres Kurses zu Quantenchemie mit VQE. Für ein tieferes Verständnis der Funktionsweise von Variationsalgorithmen wie VQE empfehlen wir den Kurs Variational Algorithm Design.
Verständnisfragen
In diesem Abschnitt haben wir eine Grundzustandsenergie aus einem Hamiltonoperator berechnet. Wie würden wir vorgehen, wenn wir dies auf die Bestimmung der Geometrie eines Moleküls anwenden wollten?
Antwort
Wir müssten Variablen für die interatomaren Abstände und die Winkel zwischen den Bindungen einführen und diese variieren. Für jede Variation dieser Größen würde ein neuer Hamiltonoperator entstehen (da die die Energie beschreibenden Operatoren von der Geometrie abhängen). Für jeden so erzeugten und auf Qubits abgebildeten Hamiltonoperator müsste eine Optimierung wie die oben beschriebene durchgeführt werden. Von all diesen konvergierten Optimierungsproblemen würde die Geometrie mit der niedrigsten Energie diejenige sein, die in der Natur vorkommt. Das ist deutlich aufwendiger als das oben Gezeigte. Eine solche Berechnung wird für das einfachste Molekül, , hier durchgeführt.
4. VQEs Beziehung zu anderen Methoden
In diesem Abschnitt betrachten wir die Vor- und Nachteile des ursprünglichen VQE-Ansatzes und zeigen seine Beziehungen zu anderen, neueren Algorithmen auf.
4.1 Die Stärken und Schwächen von VQE
Einige Stärken wurden bereits erwähnt. Dazu gehören:
- Eignung für moderne Hardware: Einige Quantenalgorithmen erfordern deutlich niedrigere Fehlerraten, die an großskalige Fehlertoleranz heranreichen. VQE nicht; es kann auf aktuellen Quantencomputern implementiert werden.
- Flache Circuits: VQE verwendet häufig relativ flache Quantenschaltkreise. Das macht VQE weniger anfällig für akkumulierte Gatterfehler und geeignet für viele Fehlerminderungstechniken. Natürlich sind die Circuits nicht immer flach; das hängt vom verwendeten Ansatz ab.
- Vielseitigkeit: VQE kann (prinzipiell) auf jedes Problem angewendet werden, das sich als Eigenwert-/Eigenvektor-Problem formulieren lässt. Es gibt jedoch viele Einschränkungen, die VQE für manche Probleme unpraktisch oder nachteilig machen. Einige davon werden im Folgenden zusammengefasst.
Einige Schwächen von VQE und Probleme, für die es unpraktisch ist, wurden oben bereits beschrieben. Dazu gehören:
- Heuristische Natur: VQE garantiert keine Konvergenz zur korrekten Grundzustandsenergie, da seine Leistung von der Wahl des Ansatzes und der Optimierungsmethoden abhängt[1-2]. Wenn ein schlechter Ansatz gewählt wird, dem die erforderliche Verschränkung für den gewünschten Grundzustand fehlt, kann kein klassischer Optimierer diesen Grundzustand erreichen.
- Potenziell viele Parameter: Ein sehr ausdrucksstarker Ansatz kann so viele Parameter haben, dass die Minimierungsiterationen sehr zeitaufwendig werden.
- Hoher Messaufwand: In VQE wird Estimator verwendet, um den Erwartungswert jedes Terms im Hamiltonoperator zu schätzen. Bei den meisten relevanten Hamiltonoperatoren gibt es Terme, die nicht gleichzeitig geschätzt werden können. Das kann VQE für große Systeme mit komplizierten Hamiltonoperatoren ressourcenintensiv machen[1].
- Auswirkungen von Rauschen: Wenn der klassische Optimierer nach einem Minimum sucht, können verrauschte Berechnungen ihn verwirren und vom wahren Minimum weglenken oder die Konvergenz verzögern. Eine mögliche Lösung hierfür ist der Einsatz modernster Fehlerminderungs- und Fehlerunterdrückungstechniken[2-3] von IBM.
- Barren Plateaus: Diese Bereiche verschwindender Gradienten[2-3] existieren auch ohne Rauschen, werden durch Rauschen aber problematischer, da die durch Rauschen verursachten Änderungen der Erwartungswerte größer sein können als die Änderungen durch die Parameteraktualisierung in diesen flachen Bereichen.
4.2 Beziehung zu anderen Ansätzen
Adapt-VQE
Der ADAPT-VQE-Algorithmus (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver) ist eine Weiterentwicklung des ursprünglichen VQE-Algorithmus, die auf verbesserte Effizienz, Genauigkeit und Skalierbarkeit bei Quantensimulationen abzielt, insbesondere in der Quantenchemie.
Der ursprüngliche VQE-Algorithmus, der in dieser Lektion beschrieben wird, verwendet einen vordefinierten, festen Ansatz zur Annäherung an den Grundzustand des Systems. In unserem Fall verwendeten wir efficient_su2 mit einer einzelnen Wiederholung und Y- sowie RZ-Rotationsgattern. Obwohl sich die Parameter in den RZ-Gattern änderten, blieben Struktur und Gatter dieses Ansatzes unverändert.
ADAPT-VQE begegnet den Einschränkungen von VQE durch adaptive Ansatzkonstruktion. Anstatt mit einem festen Ansatz zu beginnen, baut ADAPT-VQE den Ansatz dynamisch und iterativ auf. In jedem Schritt wird der Operator aus einem vordefinierten Pool (etwa fermionische Anregungsoperatoren) ausgewählt, der den größten Gradienten bezüglich der Energie aufweist. So werden nur die wirkungsvollsten Operatoren hinzugefügt, was zu einem kompakten und effizienten Ansatz führt[4-6]. Dieser Ansatz kann mehrere vorteilhafte Effekte haben:
- Reduzierte Schaltkreistiefe: Durch inkrementelles Wachstum des Ansatzes und Fokus auf notwendige Operatoren minimiert ADAPT-VQE die Gatteroperationen im Vergleich zu herkömmlichen VQE-Ansätzen[5,7].
- Verbesserte Genauigkeit: Die adaptive Natur ermöglicht es ADAPT-VQE, in jedem Schritt mehr Korrelationsenergie zu erfassen, was es besonders effektiv für stark korrelierte Systeme macht, bei denen traditionelles VQE Schwierigkeiten hat[8,9].
- Skalierbarkeit und Rauschrobustheit: Der kompakte Ansatz reduziert die Anhäufung von Gattern, den Rechenaufwand und die Anzahl der zu minimierenden Variationsparameter.
ADAPT-VQE ist jedoch nicht perfekt. In manchen Fällen kann es in lokalen Minima stecken bleiben oder verlangsamt werden, und es kann unter Überparametrisierung leiden. Außerdem kann es recht ressourcenintensiv sein, da es die Berechnung von Gradienten und die Parameteroptimierung mit vielen Gatterstrukturen erfordert.
Quantenphasenschätzung (QPE)
QPE verfolgt einen ähnlichen Zweck wie VQE, unterscheidet sich jedoch erheblich in der Implementierung. QPE erfordert fehlertolerante Quantencomputer aufgrund seiner generell tiefen Quantenschaltkreise und des hohen Kohärenzgrades, den es benötigt. Sobald QPE implementiert werden kann, wäre es präziser als VQE. Ein Weg, den Unterschied zu beschreiben, ist die Präzision als Funktion der Schaltkreistiefe. QPE erreicht die Präzision mit einer Schaltkreistiefe, die als skaliert[10]. VQE benötigt Messungen, um dieselbe Präzision zu erreichen[10,11].
Krylov, SQD, QSCI und andere in diesem Kurs
VQE hat dazu beigetragen, Quantenalgorithmen zu etablieren, die weiterhin auf klassische Computer angewiesen sind – nicht nur für den Betrieb des Quantencomputers, sondern für wesentliche Teile des Algorithmus. Mehrere solcher Algorithmen stehen im Mittelpunkt des restlichen Kurses. Hier geben wir einen kurzen Überblick über einige davon, um sie mit VQE zu vergleichen. In den folgenden Lektionen werden sie deutlich ausführlicher erklärt.
Krylov-Quantendiagonalisierung (KQD)
Krylov-Unterraummethoden sind Verfahren, um eine Matrix auf einen Unterraum zu projizieren, ihre Dimension zu reduzieren und sie handhabbar zu machen, während die wichtigsten Eigenschaften erhalten bleiben. Ein Kniff dieser Methode besteht darin, einen Unterraum zu erzeugen, der diese Eigenschaften bewahrt; die Erzeugung dieses Unterraums hängt eng mit einer etablierten Methode auf Quantencomputern zusammen, der sogenannten Trotterisierung.
Es gibt einige Varianten von Quanten-Krylov-Methoden, aber im Allgemeinen lautet der Ansatz:
- Den Quantencomputer nutzen, um durch Trotterisierung einen Unterraum (den Krylov-Unterraum) zu erzeugen
- Die interessierende Matrix auf diesen Krylov-Unterraum projizieren
- Den neuen projizierten Hamiltonoperator mit einem klassischen Computer diagonalisieren
Stichprobenbasierte Quantendiagonalisierung (SQD)
Die stichprobenbasierte Quantendiagonalisierung (SQD) ist mit der Krylov-Methode verwandt, da auch sie versucht, die Dimension einer zu diagonalisierenden Matrix zu reduzieren, während wichtige Eigenschaften erhalten bleiben. SQD geht dabei wie folgt vor:
- Mit einer anfänglichen Schätzung des Grundzustands beginnen und das System in diesem Grundzustand präparieren.
- Den Sampler verwenden, um die Bitketten, aus denen dieser Zustand besteht, zu sampeln.
- Die Sammlung von Rechenbasiszuständen des Samplers als Unterraum verwenden, auf den die interessierende Matrix projiziert wird.
- Die kleinere, projizierte Matrix mit einem klassischen Computer diagonalisieren.
Das ist mit VQE verwandt, da es klassisches und Quantencomputing für wesentliche Algorithmuskomponenten nutzt. Beide teilen auch die Anforderung, eine gute anfängliche Schätzung oder einen Ansatz zu haben. Aber die Verteilung der Arbeit zwischen klassischem und Quantencomputer in SQD ähnelt eher der der Krylov-Methode.
Tatsächlich wurden die Krylov-Methode und SQD kürzlich zur stichprobenbasierten Krylov-Quantendiagonalisierungsmethode (SKQD) kombiniert[12].
Quantenunterraum-Konfigurationswechselwirkung
Die Quantum Selected Configuration Interaction (QSCI)[13] ist ein Algorithmus, der einen angenäherten Grundzustand eines Hamiltonoperators erzeugt, indem er eine Testwellenfunktion sampelt, um die bedeutenden Rechenbasiszustände zu identifizieren und damit einen Unterraum für eine klassische Diagonalisierung zu erzeugen. Sowohl SQD als auch QSCI verwenden einen Quantencomputer, um einen reduzierten Unterraum aufzubauen. QSCIs besondere Stärke liegt in seiner Zustandspräparation, insbesondere im Kontext von Chemieproblemen. Es nutzt verschiedene Strategien wie zeitentwickelte Zustände[14] und eine Reihe chemisch inspirierter Ansätze. Durch den Fokus auf effiziente Zustandspräparation reduziert QSCI die Quantenrechenkosten für chemische Hamiltonoperatoren, während es hohe Genauigkeit beibehält und die Rauschrobustheit durch Quantenzustands-Samplingtechniken nutzt[15]. QSCI bietet außerdem eine adaptive Konstruktionstechnik, die mehr Ansätze für bessere Ergebnisse liefert.
Der Standard-Workflow von QSCI für Chemieprobleme sieht wie folgt aus:
- Den molekularen Hamiltonoperator mit einer Software deiner Wahl aufbauen (z. B. SciPy).
- Einen QSCI-Algorithmus vorbereiten, indem ein geeigneter Anfangszustand und ein chemisch inspirierter Ansatz mit einem vorausgewählten Parametersatz gewählt werden.
- Bedeutende Basiszustände sampeln und den Hamiltonoperator mit einem klassischen Computer diagonalisieren, um die Grundzustandsenergie zu erhalten.
- Häufig werden Konfigurationsrekonstruktion[16] und Symmetrie-Postselection[15] als Nachverarbeitungstechniken eingesetzt.
- Optional hat der Workflow des adaptiven QSCI eine zusätzliche Optimierungsschleife von Schritt 2 zu Schritt 3, indem mehr Ansätze mit zufälligen Anfangszuständen verwendet werden.
Verständnisfragen
Was haben VQE und alle anderen oben aufgeführten Methoden gemeinsam (außer QPE, das nicht ausführlich beschrieben wird)
Antwort
Alle verwenden einen Versuchszustand oder eine Wellenfunktion irgendeiner Art. Alle funktionieren am besten, wenn die anfängliche Schätzung für diesen Versuchszustand ausgezeichnet ist.
Eine weitere richtige Antwort ist, dass sie alle am einfachsten zu implementieren sind, wenn der Hamiltonoperator leicht zu messen ist (also in relativ wenige Gruppen kommutierender Pauli-Operatoren sortiert werden kann).
Was hat VQE mit keiner der anderen oben aufgeführten Methoden gemeinsam?
Antwort
Klassische Optimierer. Keine der anderen Methoden verwendet klassische Optimierungsalgorithmen zur Auswahl von Variationsparametern.
Referenzen
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/
[17] https://arxiv.org/abs/2412.13839