Krylov-Quanten-Diagonalisierung
In dieser Lektion zur Krylov-Quanten-Diagonalisierung (KQD) werden wir folgende Fragen beantworten:
- Was ist die Krylov-Methode im Allgemeinen?
- Warum funktioniert die Krylov-Methode und unter welchen Bedingungen?
- Welche Rolle spielt Quantencomputing dabei?
Der Quantenteil der Berechnungen basiert größtenteils auf der Arbeit in Ref [1].
Das folgende Video gibt einen Überblick über Krylov-Methoden im klassischen Computing, motiviert deren Einsatz und erklärt, wie Quantencomputing in diesem Arbeitsbereich eine Rolle spielen kann. Der nachfolgende Text bietet mehr Details und implementiert eine Krylov-Methode sowohl klassisch als auch auf einem Quantencomputer.
1. Einführung in Krylov-Methoden
Eine Krylov-Unterraum-Methode kann sich auf eine von mehreren Methoden beziehen, die auf dem sogenannten Krylov-Unterraum aufgebaut sind. Eine vollständige Übersicht dieser Methoden würde den Rahmen dieser Lektion sprengen, aber Ref [2-4] bieten wesentlich mehr Hintergrundinformationen. Hier konzentrieren wir uns darauf, was ein Krylov-Unterraum ist, wie und warum er bei Eigenwertproblemen nützlich ist, und schließlich wie er auf einem Quantencomputer implementiert werden kann.
Definition: Gegeben sei eine symmetrische, positiv semidefinite -Matrix . Der Krylov-Raum der Ordnung ist der Raum, der durch Vektoren aufgespannt wird, die durch Multiplikation höherer Potenzen der Matrix bis zu mit einem Referenzvektor entstehen.
Obwohl die obigen Vektoren das aufspannen, was wir einen Krylov-Unterraum nennen, gibt es keinen Grund anzunehmen, dass sie orthogonal sind. Häufig verwendet man einen iterativen Orthonormierungsprozess ähnlich der Gram-Schmidt-Orthogonalisierung. Hier verläuft der Prozess etwas anders, da jeder neue Vektor orthogonal zu den anderen gemacht wird, während er erzeugt wird. In diesem Zusammenhang nennt man dies Arnoldi-Iteration. Beginnend mit dem Anfangsvektor erzeugt man den nächsten Vektor und stellt dann sicher, dass dieser zweite Vektor orthogonal zum ersten ist, indem man seine Projektion auf abzieht. Das heißt
Es ist nun leicht zu sehen, dass da
Dasselbe tun wir für den nächsten Vektor und stellen sicher, dass er orthogonal zu beiden vorherigen Vektoren ist:
Wenn wir diesen Prozess für alle Vektoren wiederholen, haben wir eine vollständige Orthonormalbasis für einen Krylov-Raum. Beachte, dass der Orthogonalisierungsprozess den Nullvektor liefert, sobald , da orthogonale Vektoren notwendigerweise den gesamten Raum aufspannen. Der Prozess liefert ebenfalls den Nullvektor, wenn ein Vektor ein Eigenvektor von ist, da alle nachfolgenden Vektoren Vielfache dieses Vektors wären.
1.1 Ein einfaches Beispiel: Krylov von Hand
Gehen wir die Erzeugung eines Krylov-Unterraums an einer trivial kleinen Matrix Schritt für Schritt durch, damit wir den Prozess nachvollziehen können. Wir beginnen mit einer Anfangsmatrix , die uns interessiert:
Für dieses kleine Beispiel können wir die Eigenvektoren und Eigenwerte leicht bestimmen, sogar von Hand. Wir zeigen hier die numerische Lösung.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
Wir halten sie hier zum späteren Vergleich fest:
Wir möchten untersuchen, wie dieser Prozess funktioniert (oder scheitert), wenn wir die Dimension unseres Krylov-Unterraums erhöhen. Dazu wenden wir diesen Prozess an:
- Erzeuge einen Unterraum des vollen Vektorraums, beginnend mit einem zufällig gewählten Vektor (nenne ihn , falls er bereits normiert ist, wie oben).
- Projiziere die vollständige Matrix auf diesen Unterraum und bestimme die Eigenwerte der projizierten Matrix .
- Vergrößere den Unterraum durch Erzeugen weiterer Vektoren, indem du sicherstellst, dass sie orthonormal sind, ähnlich der Gram-Schmidt-Orthogonalisierung.
- Projiziere auf den größeren Unterraum und bestimme die Eigenwerte der resultierenden Matrix .
- Wiederhole dies, bis die Eigenwerte konvergieren (oder in diesem Spielzeugbeispiel, bis du Vektoren erzeugt hast, die den gesamten Vektorraum der ursprünglichen Matrix aufspannen).
Eine normale Implementierung der Krylov-Methode müsste das Eigenwertproblem für die auf jeden Krylov-Unterraum projizierte Matrix nicht bei jeder Aufbaustufe lösen. Du könntest den Unterraum der gewünschten Dimension konstruieren, die Matrix auf diesen Unterraum projizieren und die projizierte Matrix diagonalisieren. Das Projizieren und Diagonalisieren bei jeder Unterraumdimension geschieht hier nur zur Überprüfung der Konvergenz.
Dimension :
Wir wählen einen zufälligen Vektor, sagen wir
Falls er noch nicht normiert ist, normieren wir ihn.
Nun projizieren wir unsere Matrix auf den Unterraum dieses einen Vektors:
Dies ist unsere Projektion der Matrix auf unseren Krylov-Unterraum, wenn er nur einen einzigen Vektor enthält. Der Eigenwert dieser Matrix ist trivialerweise 4. Wir können dies als unsere nullte Näherung der Eigenwerte (in diesem Fall nur einer) von betrachten. Obwohl es eine schlechte Schätzung ist, liegt sie in der richtigen Größenordnung.
Dimension :
Wir erzeugen nun den nächsten Vektor in unserem Unterraum durch Anwendung von auf den vorherigen Vektor:
Nun subtrahieren wir die Projektion dieses Vektors auf unseren vorherigen Vektor, um Orthogonalität sicherzustellen.
Falls er noch nicht normiert ist, normieren wir ihn. In diesem Fall war der Vektor bereits normiert, also
Wir projizieren nun unsere Matrix A auf den Unterraum dieser zwei Vektoren:
Wir stehen nach wie vor vor dem Problem, die Eigenwerte dieser Matrix zu bestimmen. Aber diese Matrix ist etwas kleiner als die vollständige Matrix. Bei Problemen mit sehr großen Matrizen kann die Arbeit in diesem kleineren Unterraum sehr vorteilhaft sein.
Obwohl dies immer noch keine gute Schätzung ist, ist sie besser als die nullte Näherung. Wir werden dies noch eine weitere Iteration durchführen, um sicherzustellen, dass der Prozess klar ist. Dies unterläuft jedoch den Sinn der Methode, da wir im nächsten Schritt eine 3x3-Matrix diagonalisieren werden, was bedeutet, dass wir weder Zeit noch Rechenleistung gespart haben.
Dimension :
Wir erzeugen nun den nächsten Vektor in unserem Unterraum durch Anwendung von A auf den vorherigen Vektor:
Nun subtrahieren wir die Projektion dieses Vektors auf beide vorherigen Vektoren, um Orthogonalität sicherzustellen.
Falls er noch nicht normiert ist, normieren wir ihn. In diesem Fall war der Vektor bereits normiert, also
Wir projizieren nun unsere Matrix auf den Unterraum dieser Vektoren:
Nun bestimmen wir die Eigenwerte:
Diese Eigenwerte sind exakt die Eigenwerte der ursprünglichen Matrix . Das muss so sein, da wir unseren Krylov-Unterraum so erweitert haben, dass er den gesamten Vektorraum der ursprünglichen Matrix aufspannt.
In diesem Beispiel erscheint die Krylov-Methode möglicherweise nicht besonders einfacher als die direkte Diagonalisierung. Tatsächlich ist die Krylov-Methode, wie wir in späteren Abschnitten sehen werden, nur oberhalb einer bestimmten Matrixdimension vorteilhaft; sie soll uns helfen, Eigenwert-/Eigenvektorprobleme extrem großer Matrizen zu lösen.
Dies ist das einzige Beispiel, das wir „von Hand" durcharbeiten werden, aber Abschnitt 2 unten zeigt rechnerische Beispiele.
Klärung der Begriffe
Ein häufiges Missverständnis ist, dass es für ein gegebenes Problem nur einen einzigen Krylov-Unterraum gibt. Natürlich gibt es, da es viele Anfangsvektoren gibt, auf die unsere Matrix angewendet werden könnte, viele mögliche Krylov-Unterräume. Wir werden den Ausdruck „der Krylov-Unterraum" nur verwenden, um auf einen bereits für ein konkretes Beispiel definierten spezifischen Krylov-Unterraum zu verweisen. Bei allgemeinen Problemlösungsansätzen werden wir auf „einen Krylov-Unterraum" verweisen.
Eine letzte Klärung: Es ist zulässig, von einem „Krylov-Raum" zu sprechen. Häufig wird er als „Krylov-Unterraum" bezeichnet, weil er im Kontext der Projektion von Matrizen aus einem Ausgangsraum in einen Unterraum verwendet wird. In Übereinstimmung mit diesem Kontext werden wir ihn hier überwiegend als Unterraum bezeichnen.
Überprüfe dein Verständnis
Erkläre, warum es (a) nicht nützlich und (b) nicht möglich ist, die Dimension des Krylov-Unterraums über die Dimension der betrachteten Matrix hinaus zu erweitern.
Antwort
(a) Da wir die Vektoren beim Erzeugen orthonormieren, bildet ein Satz von solcher Vektoren eine vollständige Basis, d. h. eine Linearkombination von ihnen kann verwendet werden, um jeden Vektor im Raum zu erzeugen.
(b) Der Orthogonalisierungsprozess besteht darin, die Projektion eines neuen Vektors auf alle vorherigen Vektoren abzuziehen. Wenn alle vorherigen Vektoren den gesamten Vektorraum aufspannen, ergibt das Abziehen der Projektionen auf den vollständigen Unterraum immer den Nullvektor.
Angenommen, ein Kollege demonstriert die Krylov-Methode, angewendet auf eine kleine Spielzeugmatrix. Gibt es etwas an der Wahl der Matrix und des Anfangsvektors auszusetzen?
und
Antwort
Dein Kollege hat versehentlich einen Eigenvektor als Anfangsvektor gewählt. Die Anwendung der Matrix auf den Anfangsvektor liefert einfach denselben Vektor zurück, skaliert mit dem Eigenwert. Damit wird kein Unterraum zunehmender Dimension erzeugt. Rate deinem Kollegen, einen anderen Anfangsvektor zu wählen und dabei sicherzustellen, dass es kein Eigenvektor ist.
Wende die Krylov-Methode auf die gegebene Matrix an und wähle einen geeigneten neuen Anfangsvektor. Schreibe die Schätzungen des minimalen Eigenwerts bei der nullten und ersten Ordnung deines Krylov-Unterraums auf.
Antwort
Es gibt viele mögliche Antworten, je nach Wahl des Anfangsvektors. Wir wählen:
Um zu erhalten, wenden wir einmal auf an und machen dann orthogonal zu
Bei nullter Ordnung ist die Projektion auf unseren Krylov-Unterraum
Bei erster Ordnung ist die Projektion auf diesen Krylov-Unterraum
Dies kann von Hand berechnet werden, ist aber am einfachsten mit numpy:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
Ausgabe:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
Die Schätzung des minimalen Eigenwerts beträgt -0.414.
1.2 Arten von Krylov-Methoden
„Krylov-Unterraum-Methoden" können sich auf eine von mehreren iterativen Techniken beziehen, die zur Lösung großer linearer Systeme und Eigenwertprobleme verwendet werden. Was sie alle gemeinsam haben, ist, dass sie eine Näherungslösung aus einem Krylov-Unterraum konstruieren:
wobei der Anfangsschätzvektor ist (siehe Ref [5]). Sie unterscheiden sich darin, wie sie die beste Näherung aus diesem Unterraum auswählen, und balancieren dabei Faktoren wie Konvergenzrate, Speicherbedarf und Gesamtrechenaufwand. Der Schwerpunkt dieser Lektion liegt auf der Nutzung von Quantencomputing im Kontext von Krylov-Unterraum-Methoden; eine erschöpfende Diskussion dieser Methoden geht über ihren Rahmen hinaus. Die nachstehenden kurzen Definitionen dienen nur als Kontext und enthalten einige Verweise für eine weiterführende Untersuchung dieser Methoden.
Die konjugierte Gradientenmethode (CG-Methode): Diese Methode wird zur Lösung symmetrischer, positiv definiter linearer Systeme verwendet [6]. Sie minimiert die A-Norm des Fehlers bei jeder Iteration, was sie besonders effektiv für Systeme macht, die aus diskretisierten elliptischen PDEs entstehen [7]. Wir werden diesen Ansatz im nächsten Abschnitt verwenden, um zu motivieren, warum ein Krylov-Unterraum ein effektiver Unterraum wäre, in dem man nach verbesserten Lösungen linearer Systeme suchen könnte.
Die verallgemeinerte minimale Residualmethode (GMRES-Methode): Sie ist zur Lösung allgemeiner nichtsymmetrischer linearer Systeme konzipiert. Sie minimiert die Residualnorm über einen Krylov-Raum bei jeder Iteration, was sie robust, aber potenziell speicherintensiv für große Systeme macht [7].
Die minimale Residualmethode (MINRES-Methode): Diese Methode wird zur Lösung symmetrischer indefiniter linearer Systeme verwendet. Sie ähnelt GMRES, nutzt jedoch die Symmetrie der Matrix, um den Rechenaufwand zu reduzieren [8].
Weitere erwähnenswerte Ansätze sind die vollständige Orthogonalisierungsmethode (FOM), die eng mit der Arnoldi-Methode für Eigenwertprobleme verwandt ist, die bi-konjugierte Gradientenmethode (BiCG) und die Methode der induzierten Dimensionsreduktion (IDR).
1.3 Warum die Krylov-Unterraum-Methode funktioniert
Hier werden wir motivieren, dass die Krylov-Unterraum-Methode eine effiziente Möglichkeit sein sollte, Matrixeigenwerte durch iterative Verfeinerung von Eigenvektornäherungen zu approximieren – aus der Perspektive des steilsten Abstiegs. Wir werden argumentieren, dass der Raum der aufeinanderfolgenden Korrekturen zu einer gegebenen Anfangsschätzung des Grundzustands, der die schnellste Konvergenz liefert, genau ein Krylov-Unterraum ist. Wir verzichten auf einen strengen Konvergenzbeweis.
Angenommen, unsere interessierende Matrix ist symmetrisch und positiv definit. Das macht unser Argument am relevantesten für die oben beschriebene CG-Methode. Wir treffen keine Annahmen über Dünnbesetztheit; wir behaupten auch nicht, dass hermitesch sein muss (was sie sein müsste, wenn sie ein Hamiltonian wäre).
Typischerweise möchten wir ein Problem der Form
lösen. Man könnte sich vorstellen, dass , wobei eine Konstante ist, wie in einem Eigenwertproblem. Aber unsere Problemstellung bleibt vorerst allgemeiner.
Wir beginnen mit einem Vektor , der eine Näherungslösung ist. Obwohl es Parallelen zwischen diesem Schätzvektor und in Abschnitt 1.1 gibt, nutzen wir diese hier nicht. Unser Schätzvektor hat einen Fehler, den wir nennen:
Wir definieren auch das Residuum :
Hier verwenden wir den Großbuchstaben , um das Residuum von der Dimension unseres Krylov-Unterraums zu unterscheiden.

Wir wollen nun einen Korrekturschritt der Form
durchführen, der unsere Näherung hoffentlich verbessert. Hier ist ein noch zu bestimmender Vektor. Sei der Fehler nach der Korrektur. Dann gilt

Wir interessieren uns dafür, wie sich unser Fehler unter der Transformation durch unsere Matrix verhält. Berechnen wir also die -Norm des Fehlers:
wobei wir die Symmetrie von und die Relation verwendet haben. Hier ist eine von unabhängige Konstante. Wie in Abschnitt 1.2 erwähnt, ist die -Norm des Fehlers nicht die einzige Größe, die wir minimieren könnten, aber sie ist eine gute Wahl. Wir möchten sehen, wie diese Größe von unserer Wahl der Korrekturvektoren abhängt. Dazu definieren wir die Funktion durch
ist einfach der Fehler als Funktion der Korrektur , gemessen in der -Norm. Daher wollen wir so wählen, dass möglichst klein ist. Dazu berechnen wir den Gradienten von . Unter Verwendung der Symmetrie von ergibt sich
Der Gradient zeigt in Richtung des steilsten Anstiegs, d. h. sein Gegenteil gibt uns die Richtung, in der die Funktion am stärksten abnimmt: die Richtung des steilsten Abstiegs. Bei unserer Anfangsschätzung , wo , gilt Daher nimmt die Funktion am stärksten in der Richtung des Residuums ab. Unsere Anfangswahl würde also am meisten von der Addition des Vektors für ein Skalar profitieren.
Im nächsten Schritt wählen wir erneut einen Vektor und addieren ihn zur aktuellen Näherung. Mit demselben Argument wie zuvor wählen wir für ein Skalar . Wir setzen dies fort, sodass die -te Iteration unseres Vektors lautet:
Äquivalent dazu möchten wir den Raum, aus dem wir unsere verbesserten Schätzungen wählen, durch sukzessive Addition von , usw. aufbauen. Der -te Schätzvektor liegt in
Nun verwenden wir die Relation
und sehen, dass
Das heißt, der Raum, den wir aufbauen und der die korrekte Lösung am effizientesten approximiert, ist genau der Raum, der durch sukzessive Anwendung der Matrix auf aufgebaut wird. Ein Krylov-Unterraum ist der Raum, der durch die Vektoren der aufeinanderfolgenden Richtungen des steilsten Abstiegs aufgespannt wird.
Abschließend betonen wir noch einmal, dass wir keine numerischen Aussagen über die Skalierung dieses Ansatzes gemacht haben und auch nicht den komparativen Vorteil für dünnbesetzte Matrizen diskutiert haben. Dies dient nur dazu, die Verwendung von Krylov-Unterraum-Methoden zu motivieren und ein intuitives Verständnis für sie zu vermitteln. Wir werden nun das Verhalten dieser Methoden numerisch untersuchen.
Überprüfe dein Verständnis
Im obigen Arbeitsablauf haben wir vorgeschlagen, die -Norm des Fehlers zu minimieren. Welche anderen Größen könnte man bei der Suche nach dem Grundzustand und seinem Eigenwert in Betracht ziehen zu minimieren?
Antwort
Man könnte sich vorstellen, stattdessen den Residualvektor zu verwenden. Es könnte Fälle geben, in denen die Betrachtung des Fehlervektors selbst nützlich ist.
2. Krylov-Methoden in der klassischen Berechnung
In diesem Abschnitt implementieren wir Arnoldi-Iterationen rechnerisch, um einen Krylov-Unterraum bei der Lösung von Eigenwertproblemen nutzen zu können. Wir wenden dies zunächst auf ein kleines Beispiel an und untersuchen anschließend, wie die Rechenzeit mit der Größe der betrachteten Matrix skaliert. Ein zentraler Gedanke dabei ist, dass die Erzeugung der Vektoren, die den Krylov-Raum aufspannen, einen großen Teil der insgesamt benötigten Rechenzeit ausmacht. Der Speicherbedarf variiert je nach konkreter Krylov-Methode, doch Speicherbeschränkungen können den Einsatz traditioneller Krylov-Methoden begrenzen.
2.1 Einfaches kleines Beispiel
Bei der Konstruktion eines Krylov-Unterraums müssen wir die Vektoren in unserem Unterraum orthonormieren. Definieren wir eine Funktion, die einen bereits bekannten Vektor aus unserem Unterraum vknown (nicht notwendigerweise normiert) und einen Kandidatenvektor vnext, der dem Unterraum hinzugefügt werden soll, entgegennimmt und vnext orthogonal zu vknown sowie normiert macht. Definieren wir außerdem eine Funktion, die diesen Prozess für alle etablierten Vektoren in unserem Krylov-Unterraum durchläuft, um eine vollständig orthonormale Menge sicherzustellen.
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
Nun definieren wir eine Funktion, die einen iterativ immer größer werdenden Krylov-Unterraum aufbaut, bis der Raum der Krylov-Vektoren den vollen Raum der ursprünglichen Matrix aufspannt. Damit können wir sehen, wie gut die mit unserer Krylov-Unterraummethode erzielten Eigenwerte mit den exakten Werten übereinstimmen – als Funktion der Dimension des Krylov-Unterraums. Wichtig ist, dass unsere Funktion krylov_full_build die Krylov-Vektoren, die projizierten Hamiltonoperatoren, die Eigenwerte und die benötigte Zeit zurückgibt.
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
Wir testen dies an einer Matrix, die zwar noch recht klein ist, aber größer als etwas, das wir von Hand berechnen möchten.
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
Wir können unsere Funktionen überprüfen, indem wir sicherstellen, dass im letzten Schritt (wenn der Krylov-Raum der vollständige Vektorraum der ursprünglichen Matrix ist) die Eigenwerte der Krylov-Methode exakt mit denen der numerischen Diagonalisierung übereinstimmen:
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
Das hat geklappt. Entscheidend ist natürlich, wie gut unsere Näherung als Funktion der Dimension des Krylov-Unterraums ist. Da wir häufig daran interessiert sind, Grundzustände und andere minimale Eigenwerte zu finden (und aus weiteren, weiter unten erläuterten algebraischen Gründen), betrachten wir unsere Schätzung des kleinsten Eigenwerts als Funktion der Krylov-Unterraumdimension. Das ist:
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
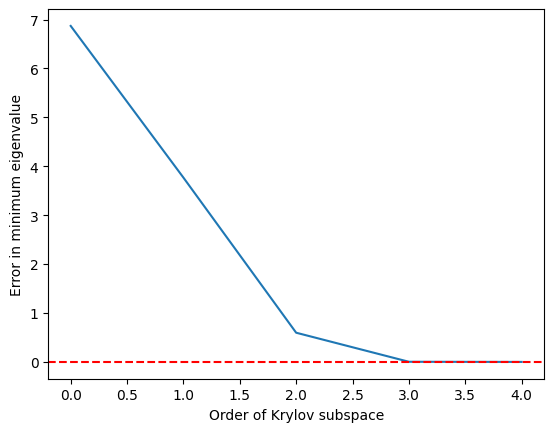
Wir sehen, dass der minimale Eigenwert recht genau erreicht wird, sobald der Krylov-Unterraum auf angewachsen ist, und bei exakt ist.
2.2 Zeitskalierung mit der Matrixdimension
Überzeugen wir uns davon, dass die Krylov-Methode gegenüber exakten numerischen Eigenwertlösern in folgender Weise vorteilhaft sein kann:
- Konstruiere Zufallsmatrizen (nicht dünn besetzt, also nicht der ideale Anwendungsfall für KQD)
- Bestimme Eigenwerte mit zwei Methoden: direkt mit NumPy und mittels eines Krylov-Unterraums.
- Wir wählen einen Grenzwert für die erforderliche Genauigkeit unserer Eigenwerte, bevor wir die Krylov-Schätzungen akzeptieren.
- Vergleiche die benötigte Wanduhrzeit für beide Methoden.
Vorbehalte: Wie wir weiter unten im Detail besprechen werden, wird Krylov-Quantendiagonalisierung am besten auf Operatoren angewendet, deren Matrixdarstellungen dünn besetzt sind und/oder mit einer kleinen Anzahl von Gruppen kommutierender Pauli-Operatoren geschrieben werden können. Die hier verwendeten Zufallsmatrizen erfüllen diese Bedingung nicht. Sie dienen lediglich dazu, die Größenordnung zu untersuchen, ab der klassische Krylov-Methoden nützlich sein könnten. Zweitens berechnen wir bei Verwendung der Krylov-Methode Eigenwerte mit vielen verschieden großen Krylov-Unterräumen. Wir geben die Zeit an, die für den minimal-dimensionalen Krylov-Unterraum benötigt wird, der unsere geforderte Genauigkeit für den Grundzustands-Eigenwert erreicht. Auch das unterscheidet sich etwas davon, ein Problem zu lösen, das für exakte Eigenwertalgorithmen unlösbar ist, da wir die exakte Lösung zur Beurteilung der benötigten Dimension heranziehen.
Wir beginnen damit, unsere Menge von Zufallsmatrizen zu erzeugen.
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
Wir diagonalisieren nun jede Matrix direkt mit numpy. Wir berechnen die benötigte Zeit für den späteren Vergleich.
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
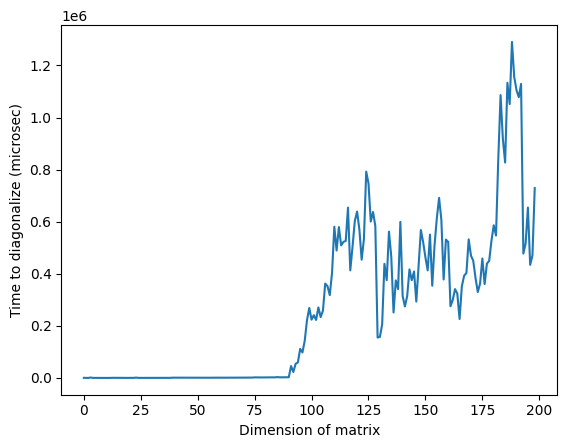
Beachte, dass die anomal hohe Zeit im obigen Bild bei einer Dimension von etwa 125 auf die zufällige Natur der Matrizen oder auf die Implementierung auf dem verwendeten klassischen Prozessor zurückzuführen sein kann, sich aber nicht reproduzieren lässt. Wird der Code erneut ausgeführt, ergibt sich ein anderes Profil mit anderen anomalen Spitzen.
Für jede Matrix bauen wir nun schrittweise einen Krylov-Unterraum auf und berechnen die Eigenwerte in Schritten. Bei jedem Schritt prüfen wir, ob der kleinste Eigenwert innerhalb unseres festgelegten absoluten Fehlers erhalten wurde. Der Unterraum, der uns erstmals Eigenwerte innerhalb des festgelegten Fehlers liefert, ist der Unterraum, für den wir die Rechenzeiten notieren. Die Ausführung dieser Zelle kann je nach Prozessorgeschwindigkeit mehrere Minuten dauern. Du kannst die Auswertung überspringen oder die maximale Dimension der diagonalisierten Matrizen reduzieren. Die Betrachtung der vorberechneten Ergebnisse ist ausreichend.
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
Stellen wir die für diese beiden Methoden ermittelten Zeiten zum Vergleich dar:
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

Dies sind die tatsächlich benötigten Zeiten. Für die Diskussion wollen wir diese Kurven jedoch glätten, indem wir über einige benachbarte Punkte bzw. Matrixdimensionen mitteln. Das geschieht im Folgenden:
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

Beachte, dass die für den Aufbau eines Krylov-Unterraums benötigte Zeit anfangs die für NumPys vollständige Diagonalisierung überschreitet. Doch mit zunehmender Matrixgröße wird die Krylov-Methode vorteilhafter. Das gilt auch, wenn wir unseren akzeptablen Fehler verringern, allerdings setzt der Vorteil dann bei einer größeren Matrixgröße ein. Es lohnt sich, das genauer zu untersuchen.
Die Zeitkomplexität der numerischen Diagonalisierung beträgt (mit gewissen Unterschieden zwischen den Algorithmen). Die Zeitkomplexität der Erzeugung einer Orthonormalbasis aus Vektoren ist ebenfalls . Der Vorteil der Krylov-Methode hängt also nicht mit der Verwendung Orthonormalbasis zusammen, sondern mit der Verwendung einer bestimmten Orthonormalbasis, die die Eigenwerte von Interesse effektiv herausgreift. Wir haben das bereits an der Skizze eines Beweises im ersten Abschnitt dieser Lektion gesehen, und das ist entscheidend für die Konvergenzgarantien in Krylov-Methoden.
Fassen wir unseren bisherigen Fortschritt zusammen:
- Für sehr große Matrizen kann die Krylov-Unterraummethode Näherungseigenwerte innerhalb geforderter Toleranzen schneller liefern als traditionelle Diagonalisierungsalgorithmen.
- Bei solch sehr großen Matrizen ist die Erzeugung eines Krylov-Unterraums der zeitaufwändigste Teil der Krylov-Unterraummethode.
- Daher wäre ein effizienter Weg zur Erzeugung eines Krylov-Unterraums von hohem Wert.
Genau hier kommt der Quantencomputer ins Spiel.
Teste dein Verständnis
Beziehe dich auf den geglätteten Plot der Diagonalisierungszeiten versus Matrixdimension weiter oben.
(a) Bei welcher Matrixdimension wurde die Krylov-Methode laut diesem Plot ungefähr schneller?
(b) Welche Aspekte der Berechnung könnten diese Dimension verschieben, ab der die Krylov-Methode schneller wird?
Antwort
(a) Die Antworten können variieren, wenn du die Berechnung erneut ausführst, aber die Krylov-Methode wird ab etwa Dimension 80–85 schneller.
(b) Es gibt viele mögliche Antworten. Einige wichtige Faktoren sind die geforderte Genauigkeit und die Dünnbesetztheit der zu diagonalisierenden Matrizen.
3. Krylov via Zeitentwicklung
Alles, was wir bisher beschrieben haben, lässt sich klassisch durchführen. Wie und wann würden wir also einen Quantencomputer einsetzen? Bei sehr großen Matrizen kann die Krylov-Methode lange Rechenzeiten und große Mengen an Arbeitsspeicher erfordern. Die Zeit, die für die Matrixoperation von auf benötigt wird, skaliert im schlimmsten Fall wie . Selbst die Multiplikation dünnbesetzter Matrizen mit einem Vektor (der typische Fall für klassische Krylov-Löser) hat eine Zeitkomplexität, die wie skaliert. Dies wird für jeden Vektor durchgeführt, den wir in unserem Unterraum haben wollen. Die Unterraumdimension ist üblicherweise kein wesentlicher Bruchteil von und skaliert oft wie . Die Erzeugung aller Vektoren skaliert also im schlimmsten Fall wie . Obwohl es weitere Schritte wie die Orthogonalisierung gibt, ist dies die dominante Skalierung, die man im Blick behalten sollte.
Das Quantencomputing erlaubt es uns, die Attribute des Problems zu verändern, die die Skalierung des Zeit- und Ressourcenbedarfs bestimmen. Anstatt einer durchgängigen Abhängigkeit von der Matrixgröße werden wir Faktoren wie die Anzahl der Shots und die Anzahl der nicht-kommutierenden Pauli-Terme, aus denen der Hamiltonoperator besteht, sehen. Lass uns untersuchen, wie das funktioniert.
3.1 Zeitentwicklung
Erinnere dich daran, dass der Operator, der einen Quantenzustand zeitlich entwickelt, ist (und es ist sehr üblich, insbesondere im Quantencomputing, das aus der Notation wegzulassen). Eine Möglichkeit, eine solche Exponentialfunktion eines Operators zu verstehen und sogar zu realisieren, besteht darin, ihre Taylor-Reihenentwicklung zu betrachten. Beachte, dass diese Operation auf einen anfänglichen Vektor eine Summe von Termen mit zunehmenden Potenzen von ergibt, die auf den Anfangszustand angewendet werden. Es sieht so aus, als könnten wir unseren Krylov-Unterraum einfach durch Zeitentwicklung unseres anfänglichen Schätzzustands erzeugen!
Der Haken liegt in der Realisierung der Zeitentwicklung auf einem echten Quantencomputer. Viele der Terme im Hamiltonoperator werden nicht miteinander kommutieren. Während also einige einfache Exponentialoperatoren wie einfachen Circuits entsprechen, gilt das für allgemeine Hamiltonoperatoren nicht. Und da sie nicht-kommutierende Terme enthalten, können wir den Exponenten nicht einfach in ein Produkt einfacher zerlegen, wie wir es bei Zahlen tun könnten.
Das ist also nicht trivial, aber es handelt sich um einen gut untersuchten Prozess im Quantencomputing. Wir führen die Zeitentwicklung auf Quantencomputern mithilfe eines Verfahrens namens Trotterisierung durch, das selbst ein reichhaltiges Thema ist[10]. Aber auf sehr hohem Niveau: Indem wir die Zeitentwicklung in sehr kleine Schritte aufteilen, zum Beispiel Schritte der Größe , begrenzen wir die Auswirkungen der Nicht-Kommutativität der Terme.
wobei .
Nennen wir einen Krylov-Unterraum der Ordnung r, den wir im klassischen Kontext direkt mithilfe von Potenzen von H erzeugt haben, einen „Power-Krylov-Unterraum".
Jetzt erzeugen wir einen ähnlichen Raum mithilfe des unitären Zeitentwicklungsoperators ; wir bezeichnen diesen als den „unitären Krylov-Raum" . Der Power-Krylov-Unterraum , den wir klassisch verwenden, kann auf einem Quantencomputer nicht direkt erzeugt werden, da kein unitärer Operator ist. Es lässt sich zeigen, dass die Verwendung des unitären Krylov-Unterraums ähnliche Konvergenzgarantien wie der Power-Krylov-Unterraum bietet: Der Grundzustandsfehler konvergiert effizient, solange der Anfangszustand einen nicht exponentiell verschwindenden Überlapp mit dem wahren Grundzustand hat und solange eine ausreichende Lücke zwischen den Eigenwerten besteht. Eine genauere Diskussion der Konvergenz findest du in Ref [1].
Hier werden Potenzen von zu verschiedenen Zeitschritten (die Potenz von entspricht einem Vorwärtsschritt um die Zeit ). Wir können das Element des Unterraums, das für die Gesamtzeit zeitentwickelt wurde, als bezeichnen.
Wir können unseren Hamiltonoperator H auf den unitären Krylov-Unterraum projizieren. Mit anderen Worten: Wir berechnen jedes Matrixelement von in der -Basis. Wir bezeichnen diese projizierte Matrix als .
3.2 Implementierung auf einem Quantencomputer
Die Matrixelemente von sind durch die Erwartungswerte gegeben, die mithilfe des Quantencomputers geschätzt werden können. Bedenke, dass als Summe von Pauli-Operatoren auf verschiedenen Qubits geschrieben werden kann und dass nicht alle Pauli-Operatoren gleichzeitig gemessen werden können. Wir können die Pauli-Terme in Gruppen kommutierender Terme sortieren und alle auf einmal messen. Aber es kann viele solcher Gruppen brauchen, um alle Terme abzudecken. Daher wird die Anzahl der verschiedenen kommutierenden Gruppen, in die die Terme partitioniert werden können, , wichtig.
Hier ist ein Pauli-String der Form oder eine Menge solcher Pauli-Strings, die miteinander kommutieren. Da wir als solche Summe messbarer Operatoren schreiben können, lassen sich die folgenden Ausdrücke für Matrixelemente von mithilfe des Qiskit Runtime Primitiv Estimator realisieren.
Dabei sind die Vektoren des unitären Krylov-Raums und die Vielfachen des gewählten Zeitschritts . Auf einem Quantencomputer kann die Berechnung jedes Matrixelements mit jedem Algorithmus durchgeführt werden, der uns den Überlapp zwischen Quantenzuständen liefert. In dieser Lektion konzentrieren wir uns auf den Hadamard-Test. Da die Dimension hat, hat der in den Unterraum projizierte Hamiltonoperator die Dimensionen . Bei einem hinreichend kleinen (im Allgemeinen reicht für eine konvergierte Schätzung der Eigenwerte) können wir den projizierten Hamiltonoperator dann leicht klassisch diagonalisieren. Wir können jedoch nicht direkt diagonalisieren, da die Krylov-Raum-Vektoren nicht orthogonal sind. Wir müssen ihre Überlappungen messen und eine Matrix konstruieren:
Dies ermöglicht es uns, das Eigenwertproblem in einem nicht-orthogonalen Raum zu lösen (auch als verallgemeinertes Eigenwertproblem bezeichnet):
Durch Betrachtung der Lösungen dieses verallgemeinerten Eigenwertproblems kann man dann Schätzungen der Eigenwerte und Eigenzustände von erhalten. Zum Beispiel erhält man die Schätzung der Grundzustandsenergie, indem man den kleinsten Eigenwert nimmt und den Grundzustand aus dem entsprechenden Eigenvektor abliest. Die Koeffizienten in bestimmen den Beitrag der verschiedenen Vektoren, die aufspannen.
Verallgemeinertes Eigenwertproblem
Warum können wir nicht einfach diagonalisieren? Da die Information über die Geometrie der Krylov-Basis enthält (die bis auf sehr spezielle Fälle nicht-orthogonal ist), beschreibt allein keine Projektion des vollständigen Hamiltonoperators, sodass seine Eigenwerte keine besondere Beziehung zu denen des vollständigen Hamiltonoperators haben – sie könnten beliebige Zufallswerte sein. Das Lösen des verallgemeinerten Eigenwertproblems ist erforderlich, um die ungefähren Eigenwerte und Eigenvektoren zu erhalten, die der Projektion des vollständigen Hamiltonoperators in den Krylov-Raum entsprechen.
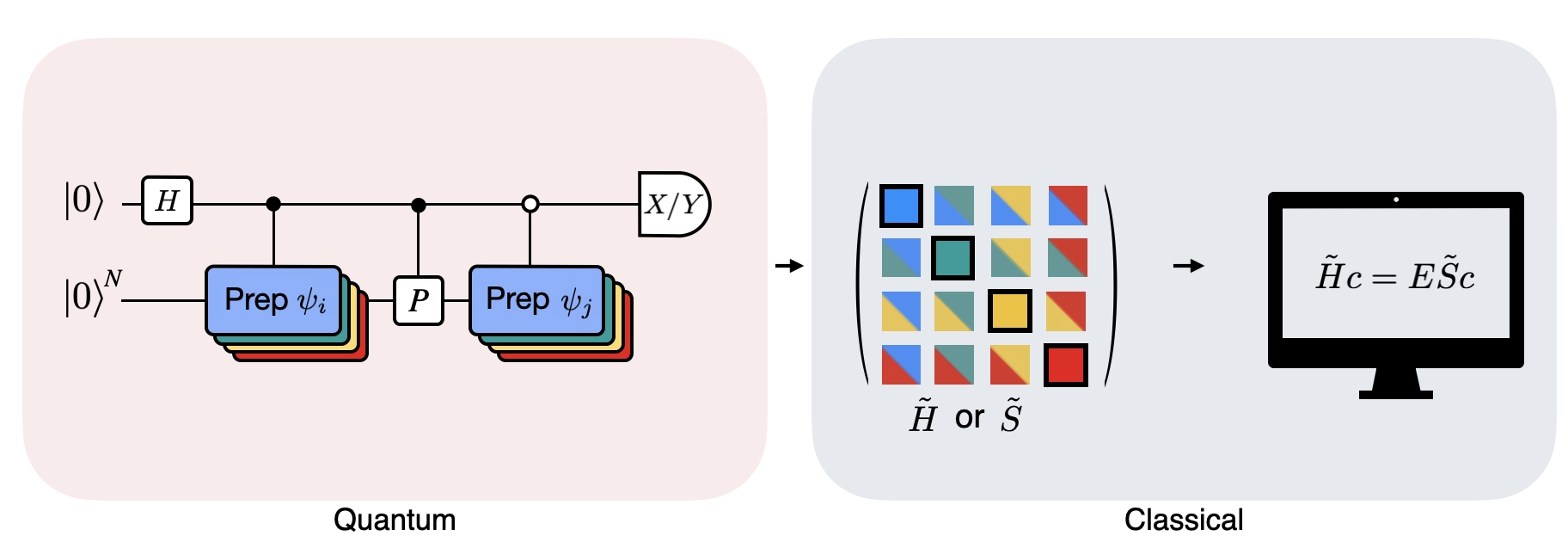
Die Abbildung zeigt eine Circuit-Darstellung des modifizierten Hadamard-Tests, einer Methode zur Berechnung des Überlapps zwischen verschiedenen Quantenzuständen. Für jedes Matrixelement wird ein Hadamard-Test zwischen den Zuständen und durchgeführt. Dies wird in der Abbildung durch das Farbschema für die Matrixelemente und die entsprechenden Operationen , hervorgehoben. Daher ist eine Reihe von Hadamard-Tests für alle möglichen Kombinationen von Krylov-Raum-Vektoren erforderlich, um alle Matrixelemente des projizierten Hamiltonoperators zu berechnen. Die obere Leitung im Hadamard-Test-Circuit ist ein Hilfs-Qubit (Ancilla), das entweder in der X- oder Y-Basis gemessen wird; sein Erwartungswert bestimmt den Wert des Überlapps zwischen den Zuständen. Die untere Leitung repräsentiert alle Qubits des System-Hamiltonoperators. Die Operation bereitet die System-Qubits im Zustand vor, kontrolliert durch den Zustand des Hilfs-Qubits (entsprechend für ), und die Operation repräsentiert die Pauli-Zerlegung des System-Hamiltonoperators . Die Implementierung auf einem Quantencomputer wird weiter unten detaillierter gezeigt.
4. Krylov-Quantendiagonalisierung auf einem Quantencomputer
Jetzt werden wir die Krylov-Quantendiagonalisierung auf einem echten Quantencomputer implementieren. Beginnen wir mit dem Import einiger nützlicher Pakete.
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
Wir definieren die folgende Funktion, um das verallgemeinerte Eigenwertproblem zu lösen, das wir gerade erläutert haben.
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
Zumindest beim anfänglichen Benchmarking ist es nützlich, eine exakte klassische Lösung zu kennen, um das Konvergenzverhalten zu überprüfen. Die folgende Funktion berechnet die Grundzustandsenergie eines Hamiltonoperators und verwendet dabei den Hamiltonoperator und die Anzahl der Qubits als Argumente.
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 Schritt 1: Problem auf Quantencircuits und Operatoren abbilden
Jetzt definieren wir einen Hamiltonoperator. Dieser unterscheidet sich von der obigen Funktion darin, dass die obige Funktion einen Hamiltonoperator als Argument nimmt und nur den Grundzustand zurückgibt, und das klassisch. Der hier definierte Hamiltonoperator bestimmt die Energieniveaus aller Energieeigenzustände und kann mithilfe von Pauli-Operatoren konstruiert und auf einem Quantencomputer implementiert werden.
Wir wählen einen Hamiltonoperator, der einer Kette von Spins entspricht, die beliebige Orientierungen im Raum annehmen können, eine sogenannte „Heisenberg-Kette". Wir nehmen an, dass der Spin von seinen nächsten Nachbarn (dem und Spin) beeinflusst werden kann, nicht aber von weiter entfernten Nachbarn. Wir berücksichtigen auch die Möglichkeit, dass die Wechselwirkung zwischen Spins unterschiedlich ist, wenn die Spins entlang verschiedener Achsen zeigen. Diese Asymmetrie entsteht manchmal, zum Beispiel aufgrund der Struktur des Kristallgitters, in das die Spins eingebettet sind.
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
Der folgende Code schränkt den Hamiltonoperator auf Einteilchenzustände ein und verwendet die Spektralnorm, um eine gute Größe für unseren Zeitschritt festzulegen. Wir wählen heuristisch einen Wert für den Zeitschritt dt (basierend auf oberen Schranken der Hamiltonoperatornorm). Ref [9] hat gezeigt, dass ein hinreichend kleiner Zeitschritt ist, und dass es bis zu einem gewissen Punkt vorzuziehen ist, diesen Wert eher zu unterschätzen als zu überschätzen, da eine Überschätzung dazu führen kann, dass Beiträge hochenergetischer Zustände sogar den optimalen Zustand im Krylov-Raum verfälschen. Andererseits führt ein zu kleiner -Wert zu einer schlechteren Konditionierung des Krylov-Unterraums, da sich die Krylov-Basisvektoren von Zeitschritt zu Zeitschritt weniger unterscheiden.
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
Wir legen die Anzahl der Trotter-Schritte fest, die bei der Zeitentwicklung verwendet werden sollen. Außerdem legen wir eine maximale Krylov-Dimension von 4 fest. Diese Krylov-Dimension ist für realistische Anwendungen nicht groß genug, aber für dieses Beispiel ausreichend. Darüber hinaus werden wir die Konvergenz bei noch kleineren Dimensionen überprüfen. In späteren Lektionen werden wir Methoden erkunden, die es uns ermöglichen, unsere Hamiltonoperatoren auf größere Unterräume zu skalieren und zu projizieren.
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
Zustandspräparation
Wähle einen Referenzzustand , der einen gewissen Überlapp mit dem Grundzustand hat. Für diesen Hamiltonoperator verwenden wir einen Zustand mit einer Anregung im mittleren Qubit als unseren Referenzzustand.
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

Zeitentwicklung
Wir können den durch einen gegebenen Hamiltonoperator erzeugten Zeitentwicklungsoperator realisieren: via der Lie-Trotter-Approximation. Der Einfachheit halber verwenden wir das eingebaute PauliEvolutionGate im Zeitentwicklungs-Circuit. Die allgemeine Syntax dafür ist folgende.
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
Wir werden eine Version davon weiter unten im Hadamard-Test verwenden, aber in Zeitschritten von vorwärtsgehen.
Hadamard-Test
Erinnere dich daran, dass wir die Matrixelemente sowohl von als auch der Gram-Matrix mithilfe des Hadamard-Tests berechnen möchten. Lass uns überprüfen, wie das in diesem Kontext funktioniert, und uns zunächst auf die Konstruktion von konzentrieren. Der Gesamtprozess ist grafisch unten dargestellt. Die Schichten farbiger Zustandspräparationsblöcke dienen als Erinnerung daran, dass dieser Prozess für alle Kombinationen von und in unserem Unterraum durchgeführt wird.
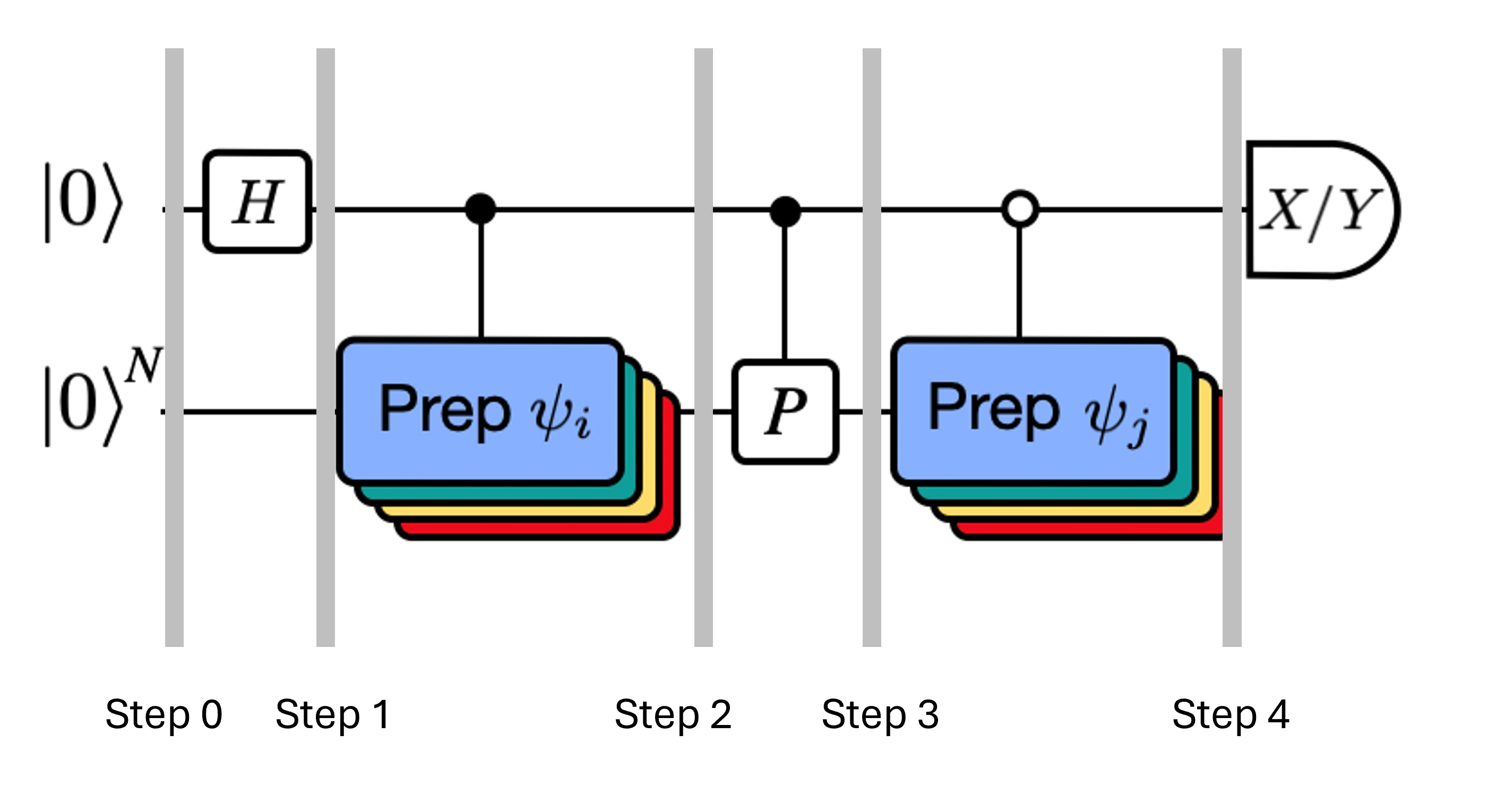
Die Zustände des Systems bei den angegebenen Schritten sind:
Hier ist ein Pauli-Term in der Zerlegung des Hamiltonoperators (beachte, dass es keine Linearkombination mehrerer kommutierender Pauli-Terme sein kann, da diese nicht unitär wäre – eine Gruppierung ist mithilfe einer anderen Konstruktion möglich, die wir später zeigen werden). , sind kontrollierte Operationen, die die Vektoren , des unitären Krylov-Raums vorbereiten, mit . Die Anwendung von Messungen von und auf diesen Circuit berechnet den Real- bzw. Imaginärteil der benötigten Matrixelemente.
Ausgehend von Schritt 4 oben wenden wir das Hadamard-Gate auf das nullte Qubit an.
Dann messen wir entweder oder .
Aus der Identität . Entsprechend liefert die Messung von :
Indem wir diese Schritte zur zuvor eingerichteten Zeitentwicklung hinzufügen, schreiben wir Folgendes.
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

Wir haben bereits auf die Tiefe von Trotter-Schaltkreisen hingewiesen. Den Hadamard-Test unter diesen Bedingungen durchzuführen, kann einen noch tieferen Schaltkreis ergeben, insbesondere nach der Zerlegung in native Gates. Das wird sich noch weiter erhöhen, wenn wir die Topologie des Geräts berücksichtigen. Deshalb ist es eine gute Idee, die 2-Qubit-Tiefe unseres Schaltkreises zu überprüfen, bevor wir Zeit auf dem Quantencomputer aufwenden.
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
Ein Schaltkreis dieser Tiefe kann auf modernen Quantencomputern keine verwertbaren Ergebnisse liefern. Wenn wir und konstruieren wollen, brauchen wir einen besseren Weg. Deshalb wird unten der effiziente Hadamard-Test eingeführt.
4.2 Schritt 2. Schaltkreise und Operatoren für die Zielhardware optimieren
Effizienter Hadamard-Test
Wir können die tiefen Schaltkreise für den Hadamard-Test, die wir erhalten haben, optimieren, indem wir einige Näherungen einführen und uns auf bestimmte Annahmen über den Modell-Hamiltonoperator stützen. Betrachte beispielsweise den folgenden Schaltkreis für den Hadamard-Test:
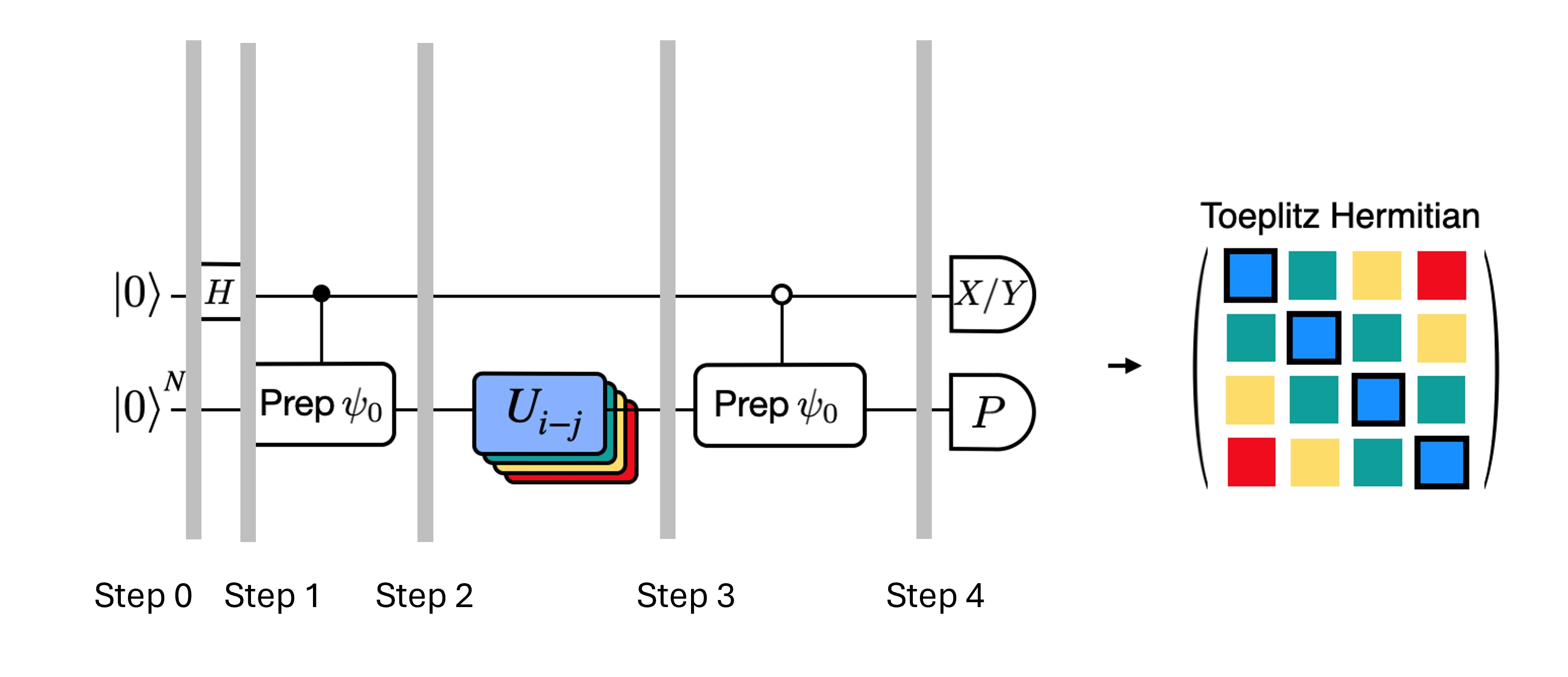
Nehmen wir an, wir können , den Eigenwert von unter dem Hamiltonoperator , klassisch berechnen. Dies ist erfüllt, wenn der Hamiltonoperator die U(1)-Symmetrie erhält. Obwohl dies wie eine starke Annahme erscheinen mag, gibt es viele Fälle, in denen es sicher ist anzunehmen, dass es einen Vakuumzustand gibt (der in diesem Fall auf den Zustand abgebildet wird), der durch die Wirkung des Hamiltonoperators unberührt bleibt. Dies gilt beispielsweise für Chemie-Hamiltonoperatoren, die stabile Moleküle beschreiben (bei denen die Anzahl der Elektronen erhalten bleibt). Da das Gate den gewünschten Referenzzustand präpariert – um beispielsweise den HF-Zustand für die Chemie zu präparieren, wäre ein Produkt aus Einzel-Qubit-NOTs, sodass kontrolliertes- nur ein Produkt aus CNOTs ist – implementiert der obige Schaltkreis folgenden Zustand vor der Messung:
wobei wir die klassisch simulierbare Phasenverschiebung von Schritt 2 zu 3 verwendet haben. Daher sind die Erwartungswerte
Mithilfe dieser Annahmen konnten wir die Erwartungswerte der interessierenden Operatoren mit weniger kontrollierten Operationen schreiben. Tatsächlich müssen wir nur die kontrollierte Zustandspräparation implementieren und keine kontrollierten Zeitentwicklungen. Durch diese Umformulierung unserer Berechnung können wir die Tiefe der resultierenden Schaltkreise erheblich reduzieren.
Zu beachten ist außerdem, dass der Pauli-Operator nun als Messung am Ende des Schaltkreises erscheint und nicht mehr als kontrolliertes Gate in der Mitte. Dadurch kann er gemeinsam mit anderen kommutierenden Pauli-Operatoren gemessen werden, wie in der Zerlegung oben angegeben.
Zeitentwicklungsoperator mit der Trotter-Zerlegung zerlegen
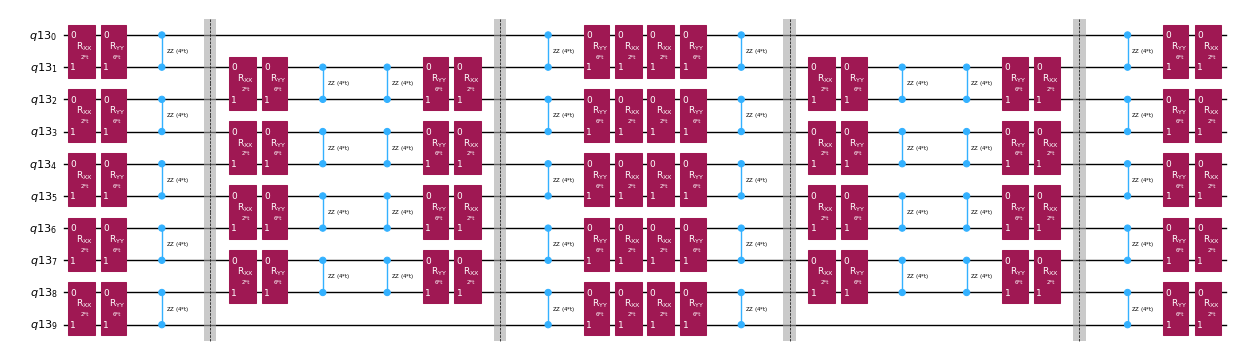
Anstatt den Zeitentwicklungsoperator exakt zu implementieren, können wir die Trotter-Zerlegung verwenden, um eine Näherung davon zu implementieren. Durch mehrfache Wiederholung einer Trotter-Zerlegung einer bestimmten Ordnung lässt sich der durch die Näherung eingeführte Fehler weiter reduzieren. Im Folgenden bauen wir die Trotter-Implementierung direkt auf die effizienteste Weise für den Wechselwirkungsgraphen des betrachteten Hamiltonoperators (nur nächste-Nachbar-Wechselwirkungen). In der Praxis fügen wir Pauli-Rotationen , , mit Kopplungsstärken und und einem parametrisierten Winkel ein, die der näherungsweisen Implementierung von entsprechen. Aufgrund des Unterschieds in der Definition der Pauli-Rotationen und der Zeitentwicklung, die wir implementieren wollen, müssen wir den Parameter verwenden, um eine Zeitentwicklung von zu erreichen. Darüber hinaus kehren wir für ungerade Wiederholungszahlen der Trotter-Schritte die Reihenfolge der Operationen um, was funktional äquivalent ist, aber erlaubt, benachbarte Operationen in einem einzigen -Unitären zusammenzufassen. Das ergibt einen wesentlich flacheren Schaltkreis als mit der generischen Funktionalität PauliEvolutionGate().
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
Wir bereiten erneut einen Anfangszustand für diesen effizienten Hadamard-Test vor.
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

Template-Schaltkreise zur Berechnung der Matrixelemente von und via Hadamard-Test
Der einzige Unterschied zwischen den Schaltkreisen, die beim Hadamard-Test verwendet werden, ist die Phase im Zeitentwicklungsoperator und die gemessenen Observablen. Deshalb können wir einen Template-Schaltkreis erstellen, der den generischen Schaltkreis für den Hadamard-Test darstellt, mit Platzhaltern für die Gates, die vom Zeitentwicklungsoperator abhängen.
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
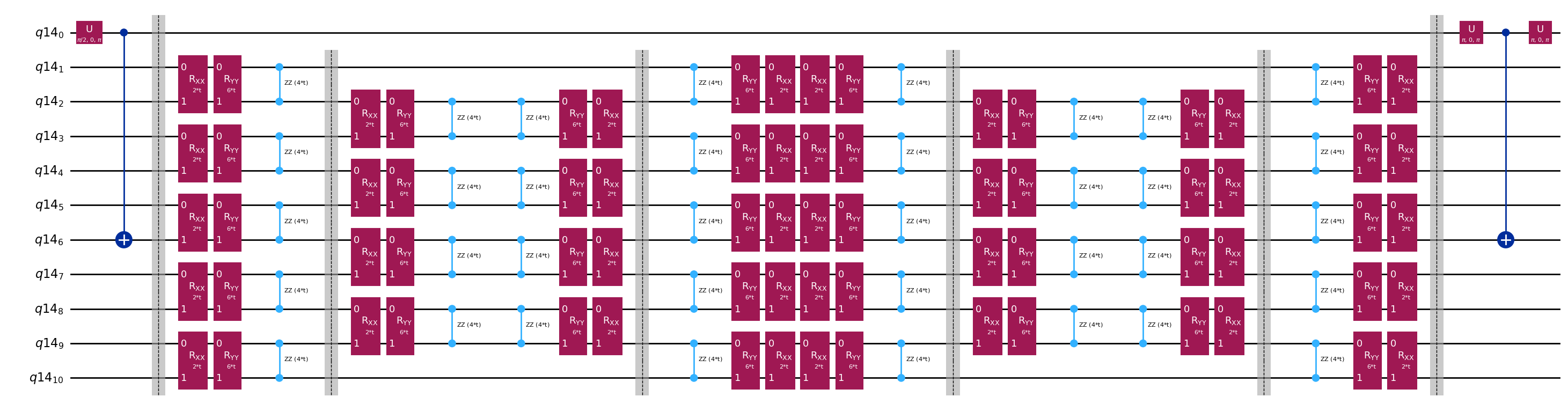
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
Diese Tiefe ist im Vergleich zum ursprünglichen Hadamard-Test erheblich reduziert. Diese Tiefe ist auf modernen Quantencomputern handhabbar, obwohl sie noch recht hoch ist. Wir werden modernste Fehlerminderung einsetzen müssen, um verwertbare Ergebnisse zu erhalten.
Wähle einen Backend, auf dem du unsere Quanten-Krylov-Berechnung ausführen möchtest, damit wir unseren Schaltkreis für diesen Quantencomputer transpilieren können.
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
Jetzt transpilieren wir unsere Schaltkreise und Operatoren.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

Nach der Optimierung ist die transpilierte Zwei-Qubit-Tiefe weiter reduziert.
4.3 Schritt 3. Mit einem Qiskit Runtime Primitive ausführen
Jetzt erstellen wir PUBs für die Ausführung mit dem Estimator.
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
Schaltkreise für sind klassisch berechenbar. Wir führen dies durch, bevor wir mit dem Fall auf einem Quantencomputer fortfahren.
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
Obwohl wir unsere Gate-Tiefe durch den effizienten Hadamard-Test um Größenordnungen reduzieren konnten, ist die Tiefe immer noch ausreichend, um modernste Fehlerminderung zu erfordern. Im Folgenden legen wir die Attribute der verwendeten Fehlerminderung fest. Alle eingesetzten Methoden sind wichtig, aber es lohnt sich, die probabilistische Fehleramplifikation (PEA) besonders hervorzuheben. Diese leistungsstarke Technik ist mit einem erheblichen Quanten-Overhead verbunden. Die hier durchgeführte Berechnung kann auf einem echten Quantencomputer 20 Minuten oder länger dauern. Du kannst mit den folgenden Parametern experimentieren, um Präzision und damit den Overhead zu erhöhen oder zu verringern. Die unten aufgeführten Standardeinstellungen liefern hochwertige Ergebnisse.
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
Schließlich führen wir die Schaltkreise für und mit dem Estimator aus.
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 Schritt 4. Nachbearbeitung und Analyse der Ergebnisse
Was wir vom Quantencomputer erhalten haben, sind die einzelnen Matrixelemente von sowie die kommutierenden Pauli-Gruppen, aus denen sich die Matrixelemente von zusammensetzen. Diese Terme müssen kombiniert werden, um unsere Matrizen zu rekonstruieren, damit wir das verallgemeinerte Eigenwertproblem lösen können.
# Store the outputs as 'results'.
results = job.result()[0]
Berechnung der effektiven Hamilton- und Überlappmatrizen
Zunächst berechnen wir die Phase, die der -Zustand während der unkontrollierten Zeitentwicklung akkumuliert:
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
Sobald wir die Ergebnisse der Schaltkreisausführungen vorliegen haben, können wir die Daten nachbearbeiten, um die Matrixelemente von zu berechnen:
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
Und die Matrixelemente von :
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
Schließlich können wir das verallgemeinerte Eigenwertproblem für lösen:
und so eine Schätzung der Grundzustandsenergie erhalten:
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
Für einen Einzel-Teilchen-Sektor können wir den Grundzustand dieses Sektors des Hamiltonians effizient klassisch berechnen:
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
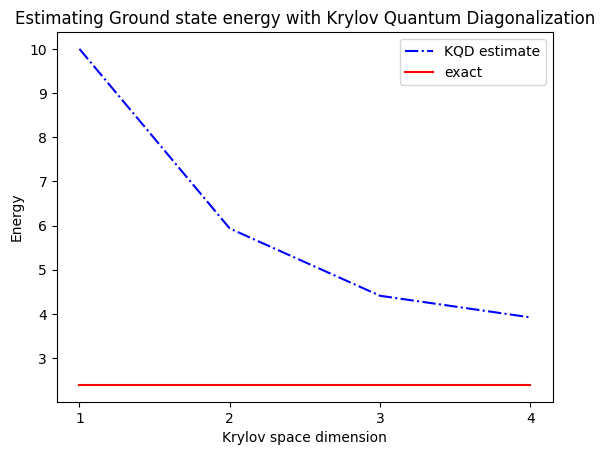
5. Diskussion und Erweiterung
Zur Zusammenfassung: Wir beginnen mit einem Referenzzustand und entwickeln ihn dann für verschiedene Zeiträume, um den unitären Krylov-Unterraum zu erzeugen. Wir projizieren unseren Hamiltonian auf diesen Unterraum. Außerdem schätzen wir die Überlappungen der Unterraum-Vektoren. Schließlich lösen wir das niedrigerdimensionale, verallgemeinerte Eigenwertproblem klassisch.
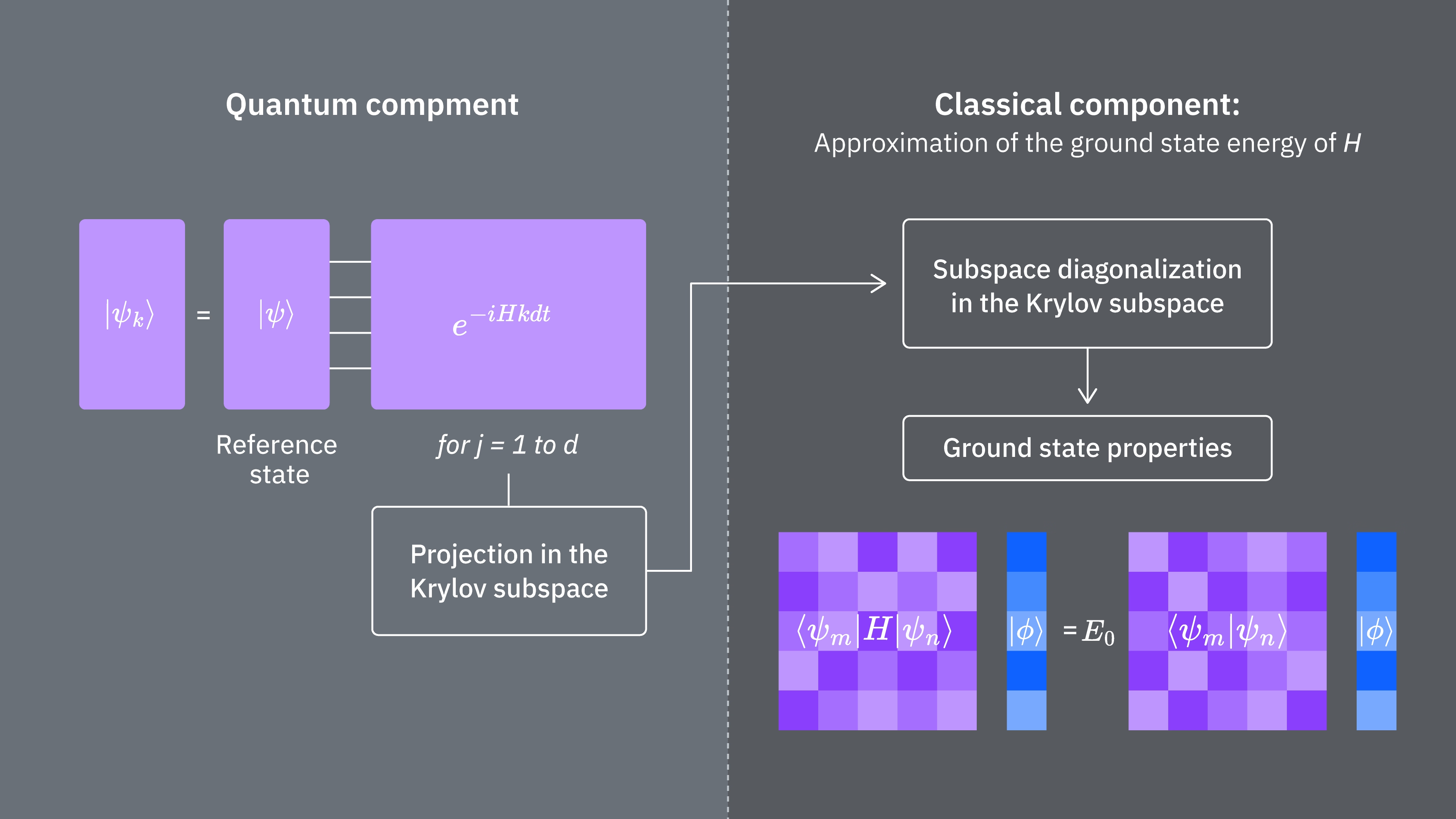
Vergleichen wir, welche Faktoren die Rechenkosten der klassischen und der quantenmechanischen Anwendung der Krylov-Methode bestimmen. Nicht alle Schritte lassen sich zwischen dem klassischen und dem quantenmechanischen Ansatz direkt vergleichen. Die folgende Tabelle fasst die Skalierung verschiedener Schritte zum Vergleich zusammen.
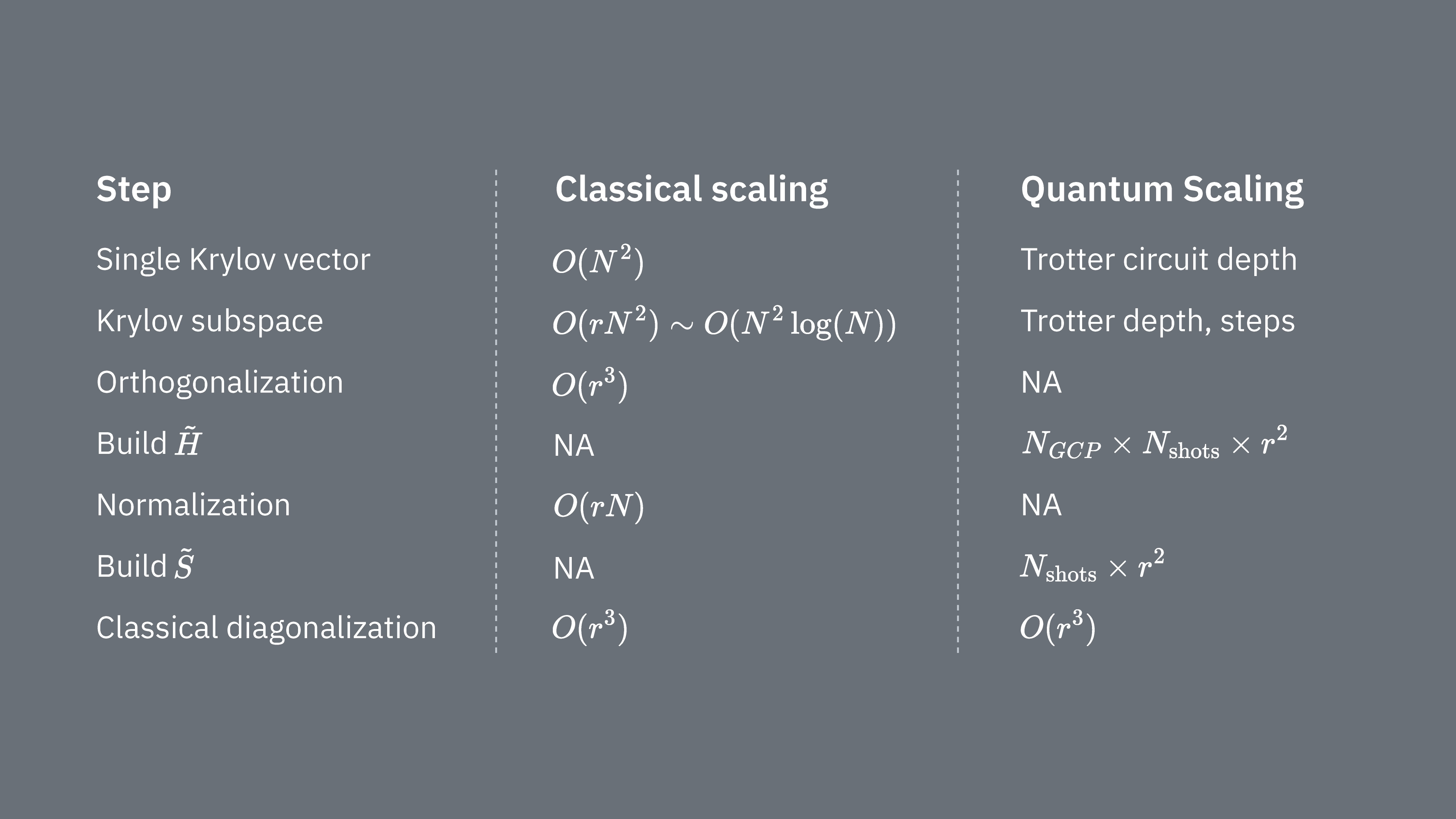
Beachte, dass Hamiltonians generell Terme enthalten, die nicht gleichzeitig gemessen werden können (da sie nicht miteinander kommutieren). Wir sortieren die Terme des Hamiltonians in Gruppen kommutierender Pauli-Operatoren, die alle gleichzeitig gemessen werden können – dabei können viele solcher Gruppen erforderlich sein, um alle nicht-kommutierenden Terme zu erfassen. Der Aufbau von auf einem Quantencomputer erfordert separate Messungen für jede Gruppe kommutierender Pauli-Strings im Hamiltonian, und jede davon benötigt viele Shots. Dies muss für verschiedene Matrixelemente durchgeführt werden, entsprechend Kombinationen verschiedener Zeitentwicklungsfaktoren. Es gibt manchmal Möglichkeiten, dies zu reduzieren, aber in dieser groben Betrachtung skaliert der Zeitaufwand dafür wie . Die Elemente von müssen geschätzt werden, was wie skaliert. Schließlich benötigt die klassische Lösung des verallgemeinerten Eigenwertproblems im projizierten Raum .
Wir sehen, dass die quantenmechanische Krylov-Diagonalisierung besonders nützlich sein kann, wenn die Anzahl der kommutierenden Pauli-Gruppen im Hamiltonian relativ gering ist. Diese Skalierungsabhängigkeiten deuten auf Anwendungsfälle hin, in denen die Krylov-Methode hilfreich ist, und auf solche, in denen sie es wahrscheinlich nicht ist. Manche Hamiltonians weisen eine hohe Komplexität auf, wenn sie auf Qubits abgebildet werden, und umfassen viele nicht-kommutierende Pauli-Strings, die sich nicht leicht in wenige kommutierende Gruppen einteilen lassen. Dies trifft zum Beispiel häufig auf Quantenchemieprobleme zu. Diese Komplexität stellt nahe Quantencomputer vor zwei wesentliche Herausforderungen:
- Die Schätzung jedes Elements von wird aufgrund der großen Anzahl von Termen rechenaufwändig.
- Die erforderlichen Trotter-Schaltkreise werden unzulässig tief.
Beide oben genannten Punkte werden weniger problematisch sein, sobald Quantencomputer Fehlertoleranz erreichen, müssen jedoch kurzfristig berücksichtigt werden. Selbst Systeme mit „einfacheren" Abbildungen als jene in der Quantenchemie können auf dieselben Hindernisse stoßen, wenn ihre Hamiltonians zu viele nicht-kommutierende Terme aufweisen. Die Krylov-Methode ist am nützlichsten, wenn der Hamiltonian in verhältnismäßig wenige kommutierende Pauli-Gruppen unterteilt werden kann und sich leicht in Trotter-Schaltkreisen implementieren lässt. Beide Bedingungen sind beispielsweise für viele Gittermodelle in der Physik erfüllt. KQD ist besonders hilfreich, wenn über den Grundzustand sehr wenig bekannt ist. Das liegt an seinen inhärenten Konvergenzgarantien und seiner Anwendbarkeit in Szenarien, in denen alternative Methoden aufgrund unzureichenden Wissens über den Grundzustand nicht in Frage kommen.
Obwohl KQD ein leistungsfähiges Werkzeug ist, bieten die zeitaufwändigen Aspekte des Protokolls – insbesondere die Schätzung jedes Elements des projizierten Hamiltonians und die Überlappung der Krylov-Zustände – Verbesserungspotenzial. Ein alternativer Ansatz verbindet Krylov-Methoden mit stichprobenbasierten Methoden, die Gegenstand der nächsten Lektion sind.
6. Anhänge
Anhang I: Krylov-Unterraum aus reellen Zeitentwicklungen
Der unitäre Krylov-Raum ist definiert als
für einen Zeitschritt , den wir später bestimmen werden. Wir nehmen vorübergehend an, dass gerade ist: dann definieren wir . Man beachte, dass der Hamiltonian bei der Projektion in den oben genannten Krylov-Raum vom Krylov-Raum
nicht unterscheidbar ist – also dem Fall, bei dem alle Zeitentwicklungen um Zeitschritte nach hinten verschoben sind. Der Grund für die Ununterscheidbarkeit liegt darin, dass die Matrixelemente
gegenüber globalen Verschiebungen der Evolutionszeit invariant sind, da die Zeitentwicklungen mit dem Hamiltonian kommutieren. Für ungerades können wir die Analyse für verwenden.
Wir wollen zeigen, dass in diesem Krylov-Raum mit Sicherheit ein energiearmer Zustand vorhanden ist. Wir tun dies mithilfe des folgenden Ergebnisses, das aus Theorem 3.1 in [3] abgeleitet ist:
Behauptung 1: Es existiert eine Funktion , sodass für Energien im Spektralbereich des Hamiltonians (d. h. zwischen der Grundzustandsenergie und der maximalen Energie) gilt ...
- für alle Werte von , die von entfernt liegen, d. h. exponentiell unterdrückt sind
- ist eine Linearkombination von für
Wir geben unten einen Beweis an, der jedoch bedenkenlos übersprungen werden kann, sofern man kein Interesse an der vollständigen, rigorosen Argumentation hat. Zunächst konzentrieren wir uns auf die Implikationen der obigen Behauptung. Aus Eigenschaft 3 können wir erkennen, dass der oben verschobene Krylov-Raum den Zustand enthält. Dies ist unser energiearmer Zustand. Um zu verstehen, warum, schreiben wir in der Energieeigenbasis:
wobei der -te Energieeigenzustand und seine Amplitude im Anfangszustand ist. Ausgedrückt in dieser Basis ergibt sich zu
wobei wir ausnutzen, dass wir durch ersetzen können, wenn es auf den Eigenzustand wirkt. Der Energiefehler dieses Zustands ist daher
Um daraus eine leichter verständliche obere Schranke zu gewinnen, teilen wir die Summe im Zähler zunächst in Terme mit und Terme mit auf:
Den ersten Term können wir durch nach oben abschätzen:
wobei der erste Schritt daraus folgt, dass für jedes in der Summe gilt, und der zweite Schritt daraus folgt, dass die Summe im Zähler eine Teilmenge der Summe im Nenner ist. Für den zweiten Term schätzen wir den Nenner nach unten durch ab, da : alles zusammenaddiert ergibt sich
Um das Verbleibende zu vereinfachen, beachte, dass für all diese aus der Definition von folgt, dass . Indem wir zusätzlich und nach oben abschätzen, ergibt sich
Dies gilt für jedes : setzen wir gleich unserem Ziel-Fehler, so konvergiert die obige Fehlergrenze exponentiell mit der Krylov-Dimension gegen diesen Wert. Außerdem entfällt der -Term in der obigen Schranke vollständig, wenn gilt.
Um die Argumentation zu vervollständigen, sei zunächst darauf hingewiesen, dass das Obige lediglich der Energiefehler des speziellen Zustands ist, nicht der Energiefehler des Zustands mit der niedrigsten Energie im Krylov-Raum. Durch das (Rayleigh-Ritz-)Variationsprinzip ist der Energiefehler des Zustands mit der niedrigsten Energie im Krylov-Raum jedoch nach oben durch den Energiefehler eines beliebigen Zustands in diesem Krylov-Raum beschränkt. Das Obige stellt daher auch eine obere Schranke für den Energiefehler des Zustands mit der niedrigsten Energie dar, d. h. für die Ausgabe des Krylov-Quanten-Diagonalisierungsalgorithmus.
Eine ähnliche Analyse wie die obige kann durchgeführt werden, die zusätzlich Rauschen und das im Notebook besprochene Schwellenwertverfahren berücksichtigt. Siehe [2] und [4] für diese Analyse.
Anhang II: Beweis von Behauptung 1
Das Folgende ist größtenteils aus [3], Theorem 3.1, abgeleitet: Sei und sei der Raum der Residualpolynome (Polynome, deren Wert an der Stelle 0 gleich 1 ist) vom Grad höchstens . Die Lösung von
lautet
und der entsprechende Minimalwert ist
Wir wollen dies in eine Funktion umwandeln, die sich natürlich durch komplexe Exponentialfunktionen ausdrücken lässt, da diese die reellen Zeitentwicklungen sind, die den quantenmechanischen Krylov-Raum erzeugen. Dazu ist es hilfreich, die folgende Transformation einzuführen, die Energien innerhalb des Spektralbereichs des Hamiltonians auf Zahlen im Bereich abbildet: wir definieren
wobei ein Zeitschritt ist, sodass gilt. Man beachte, dass und mit wachsendem Abstand von zunimmt.
Unter Verwendung des Polynoms mit den Parametern , und definieren wir die Funktion:
wobei die Grundzustandsenergie ist. Durch Einsetzen von erkennen wir, dass ein trigonometrisches Polynom vom Grad ist, d. h. eine Linearkombination von für . Aus der Definition von oben ergibt sich ferner, dass und für jedes im Spektralbereich mit gilt
Referenzen:
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).