Abbildung (Mapping)
Schau dir das Video über Mapping von Olivia Lanes an, oder öffne es in einem separaten Fenster auf YouTube.
Einleitung
In dieser Lektion konzentrieren wir uns auf den ersten und oft schwierigsten Schritt bei der Definition eines Quantenprogramms: ein Problem auf einen Quantencomputer abzubilden. Dieser Schritt beschreibt, wie ein Nutzer mit einem Rechenproblem beginnt und es in etwas übersetzt, das auf einem Quantencomputer gelöst werden kann.
In den Lektionen 2 und 3 dieses Kurses haben wir erwähnt, dass die Mapping-Phase der erste von vier Gesamtschritten im Qiskit-Patterns-Framework ist. Aus diesen Lektionen erinnerst du dich vielleicht daran, dass das Ziel des Mappings darin besteht, ein Rechenproblem in eine Kostenfunktion oder einen Erwartungswert zu übersetzen oder umzuschreiben, den wir mit einem Quantencomputer auswerten können.
In Lektion 3 haben wir ein konkretes Beispiel mit max-cut besprochen, einem rechnerisch schwierigen, aber sehr häufigen Problem in der kombinatorischen Optimierung. In diesem Beispiel haben wir mehrere Schritte durchlaufen, um das anfängliche Graphenproblem in eines zu übersetzen, das auf einem Quantencomputer gelöst werden kann. Wir haben das Problem, die maximale Anzahl von Schnitten im Graphen zu finden, in eine Kostenfunktion transformiert, diese Kostenfunktion als Hamiltonianer umgeschrieben und dann einen Test-Quantenzustand vorbereitet, dessen Grundzustand dem maximalen Schnitt entspricht. Schließlich haben wir einen Quantencircuit konstruiert, der den Testzustand von Interesse darstellt, und dann die spezifischen Gates hinzugefügt, um die zeitliche Entwicklung des Zustands zu ermöglichen. Diese Folge von Schritten war alles Teil des Mappings. Während die genauen Schritte einzigartig für das max-cut-Problem waren, kann das gleiche allgemeine Verfahren auf viele andere Anwendungen angewendet werden, wie Quantenchemie und Quantensimulationen.
Das Mapping kann schwierig sein. Es gibt keine Einheitsstrategie für jedes einzelne Problem, daher kann es einschüchternd sein. In dieser Lektion werden wir uns einige allgemeine Überlegungen zum Mapping ansehen und dann in einige repräsentative Beispielprobleme eintauchen, um die verschiedenen Möglichkeiten zu demonstrieren, ein Problem auf einen Quantencomputer abzubilden.
Allgemeine Überlegungen
Während die genaue Strategie, die man verwendet, um ein Problem auf einen Quantencomputer abzubilden, von dem jeweiligen Problem abhängt, erfüllen sie im Allgemeinen ein paar Hauptdinge.
Zunächst musst du das Problem möglicherweise vereinfachen, um es durchführbar zu machen. Dies ist nicht einzigartig für Quantencomputing – alle wissenschaftlichen Disziplinen verwenden vereinfachte Modelle, um Phänomene zu untersuchen, die sie interessieren, während sie irrelevante Details ignorieren. In der Physik gibt es einen berühmten Ausdruck, der dieses Prinzip auf die Spitze treibt: „Nehmen wir an, die Kuh ist eine Kugel." Es ist oft zu schwierig, ein System genau so zu beschreiben, wie es erscheint, aber wir können stattdessen vernünftige Vereinfachungen vornehmen, die dennoch zu hilfreichen Lösungen führen können. Einige Beispiele, wie wir das im Quantencomputing tun könnten, sind die Begrenzung der Größe oder der Circuit-Tiefe durch die Wahl eines hardware-effizienten Ansatzes, das Abschneiden komplexer Zeitentwicklungen oder das Vernachlässigen von Hamiltonianer-Termen, die wenig zur endgültigen Energie eines Quantenzustands beitragen.
Zweitens beinhaltet das Mapping das Schreiben des Problems auf eine Weise, die ein Quantencomputer verstehen kann. Das beinhaltet oft das Stellen dieser drei Fragen:
- Was werden unsere Qubits in unserem Modell darstellen?
- Ist unser Problem kontinuierlich? Da Quantencomputer digital sind, müssen wir, wenn das Problem kontinuierlich ist, einen Weg finden, es zu diskretisieren.
- Schließlich: Stimmt die Topologie des Problems mit der Topologie der Hardware überein? Falls nicht, müssen wir möglicherweise einige Tricks implementieren, damit es funktioniert.
Lass uns die erste Frage angehen, die im Mittelpunkt vieler Schwierigkeiten beim Verstehen von Mapping steht: Was kann ein Qubit darstellen?
Qubits können verwendet werden, um viele Dinge darzustellen. Das Erste und vielleicht Einfachste ist ein Knoten in einem Graphen. Graphen werden verwendet, um Konnektivität in vielen verschiedenen Arten von mathematischen Problemen zu zeigen, und die Knoten sind ein grundlegendes Element, das einen Punkt oder eine Entität in einem Netzwerk darstellt. Je nachdem, was das gesamte Netzwerk darstellt, könnte das eine Stadt, eine Person oder ein Ferromagnet in einem Gitter sein.
Qubits können auch verwendet werden, um Bosonen und Fermionen darzustellen, obwohl ich hier warnen möchte, dass ein einzelnes Qubit nicht genau einem Boson oder einem Fermion entspricht – es ist etwas komplizierter als das, wie wir in der Lektion noch besprechen werden.
Nun werden die Beispiele etwas komplizierter. Bei diesen Modellen macht es keinen Sinn mehr, von einzelnen Qubits zu sprechen; stattdessen brauchen wir Gruppen von ihnen, um etwas Physisches zu ergeben. Zum Beispiel kann eine Gruppe von Qubits, hier auf einer Heavy-Hex-Topologie dargestellt, verwendet werden, um geometrische Positionen von Aminosäuren darzustellen; Polymerketten. Ein weiteres Beispiel ist die Simulation von Hadronen-Streuung in Hochenergiephysikmodellen, die durch Simulation der zeitlichen Entwicklung eines hadronischen Wellenpakets durchgeführt werden kann. In diesem Fall kann ein Register von Qubits verwendet werden, um verschiedene Zustände eines Quantenfeldes zu kodieren; den Vakuumzustand dieses Feldes oder ein Wellenpaket, das sich auf diesem Vakuum ausbreitet.
Aber an diesem Punkt haben wir abstrakt genug über die vor uns liegende Herausforderung gesprochen. Schauen wir uns diese Beispiele im Detail an.
Mapping-Beispiele
Max-cut
Beginnen wir mit unserem ersten Beispiel. Eines der einfachsten Mapping-Probleme ist das, das wir bereits eingehend behandelt haben: das max-cut-Beispiel. Bei diesem Problem war das Mapping für uns relativ einfach, da ein Qubit einem Knoten in unserem Graphen entsprach.
Erinnere dich daran: Um das max-cut-Problem abzubilden, haben wir die Kostenfunktion mithilfe der QUBO-Formulierung als Hamiltonianer ausgedrückt. Eine Hamiltonianer-Kostenfunktion ist eine Funktion, die die optimale Lösung des Problems im Grundzustand des Hamiltonianers kodiert. Um den Kosten-Hamiltonianer zu konstruieren, haben wir die SparsePauliOp-Klasse in Qiskit verwendet, um die Konnektivität unseres Graphen anzugeben, und die Mapping-Phase zu den Quantenoperatoren war erledigt. Und der Quantencircuit war einfach der QAOA-Ansatz. Zur Auffrischung, siehe die Lektion Utility-Scale QAOA, wo wir das alles in viel mehr Detail durchgehen.
In dieser Lektion stimmte die Graphkonnektivität in dem 100-Qubit-Beispiel im Utility-Maßstab bereits mit der Topologie unserer supraleitenden Hardware überein. Wir mussten uns also nicht darum kümmern, wie wir mit verschiedenen Topologien umgehen. Aber das ist nicht immer der Fall. Wenn wir einen komplizierteren oder dichter verbundenen Graphen als die bisher hervorgehobenen Beispiele hätten, müssten wir eine Reihe von SWAP-Gates implementieren, um die effektive Konnektivität der Hardware zu ändern. Dies wird beim zweiten Schritt der Qiskit Patterns, der Transpilierung, behandelt, sollte aber schon beim Mapping-Schritt im Hinterkopf behalten werden.
Proteinfaltung
Als nächstes wollen wir uns ein Beispiel ansehen, das im Paper „Resource-efficient quantum algorithm for protein folding" modelliert wurde, das von IBM® und Mitarbeitern der University of New South Wales geschrieben wurde.
Ein wenig biochemischer Hintergrund: Proteine sind Makromoleküle, die aus langen Aminosäureketten bestehen. Diese Ketten falten sich in komplexe Strukturen, die eine Vielzahl biologischer Funktionen ausführen. Die Bestimmung der Proteinstruktur im dreidimensionalen Raum und das Verständnis der Beziehungen zwischen Struktur und Funktion gehören zu den herausforderndsten Problemen in der heutigen Biochemie. Proteine falten sich in nützliche Strukturen aufgrund von Wechselwirkungen zwischen Aminosäuren. Wenn sich eine Struktur dreht und faltet, können Aminosäuren, die entlang der Kette weit voneinander entfernt sind, direkt nebeneinander zu liegen kommen und stark wechselwirken.
Um dies auf einem Quantencomputer zu modellieren, brauchen wir einen Hamiltonianer, der alle diese Wechselwirkungen zwischen Aminosäuren beschreibt. Dann können wir die endgültige Struktur vorhersagen, indem wir den Zustand finden, der die Energie unseres Hamiltonianers minimiert. Wir werden uns hier darauf konzentrieren, wie Aminosäureketten auf einem Quantencomputer modelliert werden können und wie wir Inter-Aminosäure-Abstände zur Berechnung von Wechselwirkungsenergien erhalten können. Damit haben wir alle notwendigen Beiträge zum Hamiltonianer gesammelt, die notwendig sind, um ihn auf einem Quantencomputer zu simulieren.
In realen Proteinen können Aminosäuren ein Kontinuum möglicher Positionen einnehmen. Wir werden jedoch eine Vereinfachung verwenden und sie mit einem Gittermodell einschränken, bei dem jede Aminosäure einen Punkt auf einem Gitter einnimmt. Hier verwendeten die Autoren ein tetraedrisches Gitter. Kurze Anmerkung: Hier kodieren wir die Richtung der Kanten, nicht die Knoten selbst wie beim max-cut-Problem. Jedes Qubit stellt einen möglichen Einzelschrittweg entlang des tetraedrischen Gitters dar. Beachte, dass benachbarte Gitterplätze als A oder B bezeichnet wurden, wegen ihrer unterschiedlichen Ausrichtungen im Gitter.
Die Proteinkette wird als eine Reihe von Wendungen oder Richtungen auf diesem Gitter dargestellt. Jede Wendung zwischen Aminosäuren kann in eine von vier Richtungen verlaufen, entsprechend den Kanten des Tetraeders. Diese vier möglichen Wendungen werden mit vier Qubits in die Zustände 0001, 0010, 0100 oder 1000 kodiert.
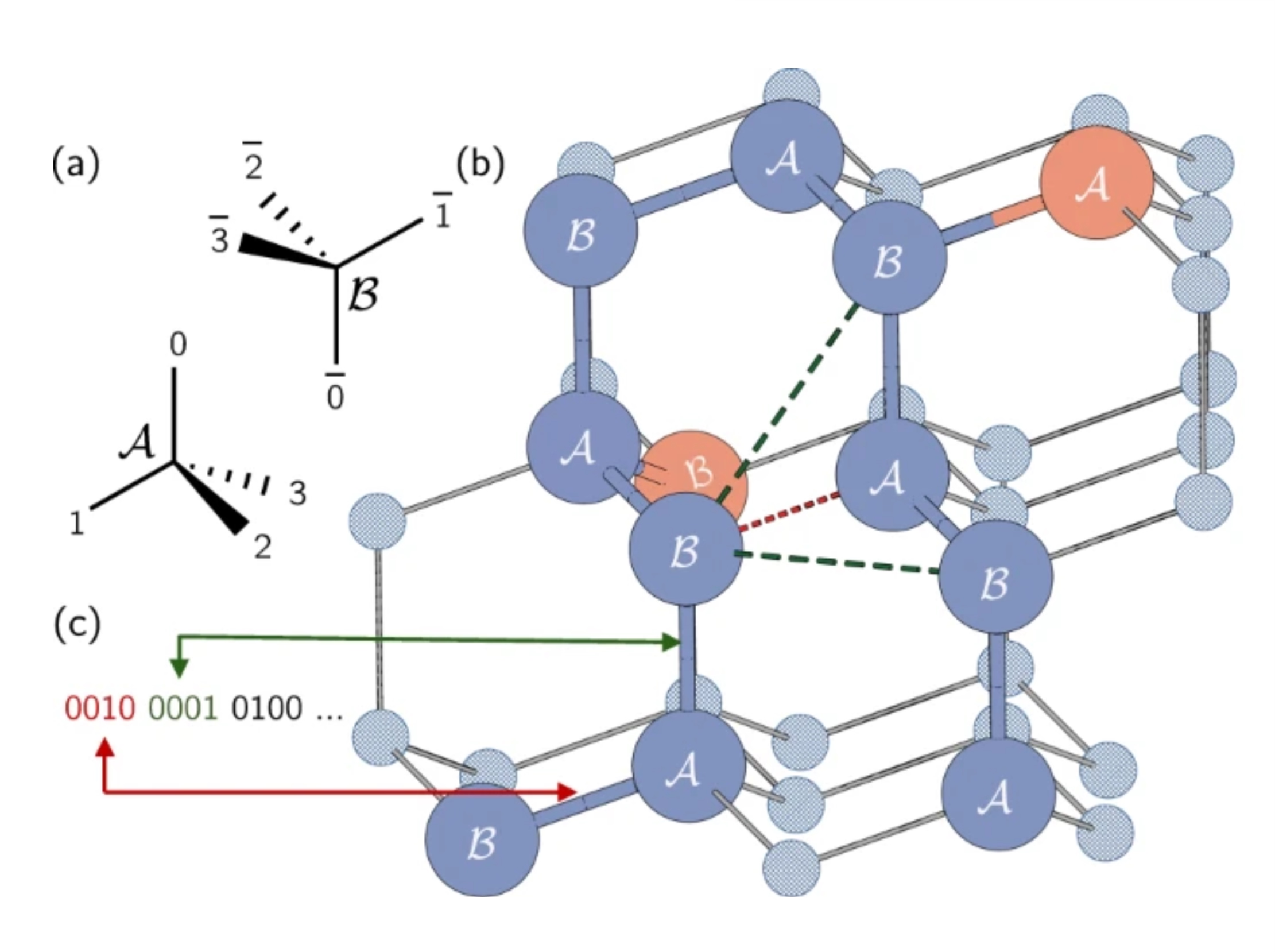
Schauen wir uns ein Beispiel in der obigen Abbildung an. Platzieren wir unsere erste Aminosäure auf dem Punkt mit der Bezeichnung „B", der in unserem tetraedrischen Gitter rot eingekreist ist. Die Richtung zur ersten Aminosäure zur zweiten ist beliebig, da das System immer so gedreht werden kann, dass diese Kante in eine beliebige Richtung zeigt. So können wir unsere zweite Aminosäure auf dem Punkt unter der ersten mit der Bezeichnung „A" platzieren. Es ist nicht so leicht zu sehen, aber der Weg von der zweiten zur dritten ist auch beliebig. Alle drei Entscheidungen würden dazu führen, dass wir zwei Kanten mit einem Winkel von ungefähr 109,5 Grad zwischen ihnen haben. Die Auswahl dieser zweiten Kante bestimmt einfach die Ausrichtung unseres Proteins im Raum. Ohne Einschränkung der Allgemeinheit können wir also die ersten beiden Wendungen einfach als 0001 und 0010 wählen.
Mit diesen Vereinfachungen wird die Konfiguration der Aminosäurekette durch diesen Ausdruck gegeben:
Bisher haben wir die Tetraeder-Kanten auf Qubits abgebildet, und unser Quantencircuit wird ein Ansatz sein. Nun müssen wir überlegen, wie wir die Energie des Problems in einen Hamiltonianer kodieren, sodass sein Grundzustand uns das optimale Faltungsmuster ergibt.
Für jede gegebene Konfiguration gibt es eine zugehörige Energie aufgrund von Wechselwirkungen zwischen den Aminosäuren. Diese Wechselwirkungen sind am stärksten, wenn die beiden Aminosäuren nahe beieinander sind. Offensichtlich werden Aminosäuren, die im Rückgrat der Kette benachbart sind, immer miteinander wechselwirken. Aber weil das Protein sich drehen und falten kann, können auch andere Aminosäurepaare wechselwirken. Aminosäuren 10 und 20 könnten beispielsweise nach dem Falten des Proteins an benachbarten Stellen enden. Wir brauchen also eine Formel, um den Abstand zwischen den Aminosäuren und mithilfe der auf den Konfigurations-Qubits kodierten Informationen zu beschreiben. Auf diese Weise können wir ihren Trennungsabstand verwenden, um zu bestimmen, wie stark sie wechselwirken.
Zunächst stellen wir eine Funktion vor, die angibt, ob eine Kante für die Wendung bei Aminosäure verwendet wird oder nicht. Hier kann eine der vier möglichen Richtungen annehmen. Basierend auf der Konfiguration, mit der wir oben begonnen haben, können wir diese Funktionen schreiben:
Dann können wir einen Unterschied in der Anzahl der -markierten Wendungen auf A- und B-Gittern vom Index bis zum Index als definieren:
Warum würden wir das tun? Nun, es stellt sich heraus, dass eine Wendung von von Gitterstelle A nach B genau durch eine Wendung von von Gitterstelle B nach A aufgehoben wird. Um also den Abstand der Aminosäure an Stelle von der an Stelle zu kennen, müssen wir nur die Differenz zwischen Schritten auf -Kanten von A-Stellen und B-Stellen finden. Da A- und B-Stellen notwendigerweise entlang des Proteinrückgrats abwechseln, wird dies durch das erfasst. Das gleiche Argument gilt für alle vier Kantentypen. Der Gesamtabstand zwischen Aminosäuren in tetraedrischen Gitterschritten kann also durch diesen Ausdruck berechnet werden:
Aber wie bekommen wir den Hamiltonianer aus dieser langen Gleichung für den Gesamtabstand zwischen den Aminosäuren? Zunächst können wir mit einfacher Geometrie vom Abstand in Gitterschritten zum Euklidischen Raum konvertieren:
Diese Abstände werden dann zur Berechnung der Energie der Proteinkonfiguration verwendet. Je nach unseren Zwecken könnten wir einen Grenzabstand festlegen, unterhalb dessen wir das Paar als wechselwirkend betrachten, oder wir könnten etwas Komplizierteres tun.
Es mag nicht offensichtlich sein, aber wir sind eigentlich mit der Mapping-Phase fertig, indem wir das tun. Die Zustände der Qubits geben die „Wendung" des Proteins an jedem Gitterpunkt an, und die Sammlung von Wendungen bestimmt den Abstand zwischen jedem Paar von Aminosäuren. Paare verschiedener Arten von Aminosäuren haben unterschiedliche Wechselwirkungen, einige attraktiv, andere abstoßend. Wenn du die Konfiguration und die Abstände nur dazu verwendest zu bestimmen, ob bekannte Aminosäure-Wechselwirkungen „ein" oder „aus" sind, wurden die Stärken dieser Wechselwirkungen bereits herausgearbeitet und können einfach in einer Tabelle wie dieser nachgeschlagen werden:
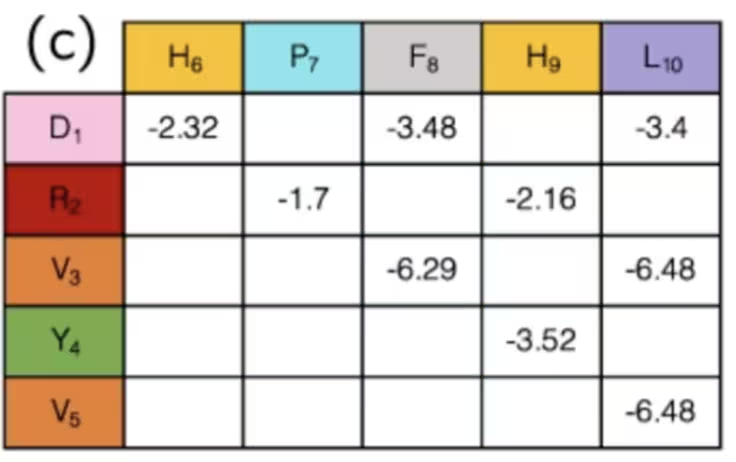
Zusammenfassend: In diesem Beispiel werden die Qubits verwendet, um Schritte auf einem Pfad entlang eines Gitters zu markieren, die zusammen eine Aminosäurekette bilden. Indem wir simulieren, wie sie sich biegen und falten, können wir hoffentlich bessere Ergebnisse in der medizinischen Forschung erzielen. Wir haben übersprungen, wie man einige Terme dieses Hamiltonianers berechnet, da sie hyper-spezifisch für dieses Problem waren, während das Definieren von Richtungen auf einem Gitter allgemeiner angewendet werden kann. Wenn du einmal einen allgemeinen Hamiltonianer hast, möchtest du ihn immer in Pauli-Operatoren übersetzen, die nativ für den Quantencomputer sind. Das werden wir als nächstes besprechen.
Jordan-Wigner-Transformation
Lass uns nun erkunden, wie man ein System subatomarer Teilchen in Pauli-Operatoren übersetzt.
Subatomare Teilchen werden in zwei Kategorien unterteilt – Bosonen und Fermionen. Bosonen, wie Photonen oder das Higgs, gehorchen einem bestimmten Satz statistischer Regeln. Fermionen, wie Elektronen oder Neutrinos, gehorchen einem anderen. Der wesentliche Unterschied zwischen ihnen ist, dass Bosonen denselben Zustand einnehmen dürfen – es gibt keine Begrenzung dafür, wie viele Bosonen sich im Grundzustand oder einem angeregten Zustand befinden können. Fermionen hingegen sind „egoistisch" – sie fordern, dass jedes einzelne Teilchen seinen eigenen Quantenzustand hat.
Bosonen haben auch ganzzahlige Spins, während Fermionen halbzahlige Spins haben, wie das Spin-1/2-Elektron und exotischere Spin-3/2-Teilchen. Um ein System von Teilchen zu beschreiben, brauchen wir eine Beschreibung ihrer Energie. Konzentrieren wir uns auf Fermionen. Wir können mit einem Hamiltonianer beginnen, der in Termen dessen geschrieben ist, was wir c-Operatoren nennen. Das sind im Grunde mathematische Objekte, die dem Erzeugen oder Vernichten eines Fermions aus einem Zustand im System entsprechen. Diese werden oft als und geschrieben, wobei der Operator ist, der ein Fermion im Zustand erzeugt, und der Operator ist, der ein Fermion im Zustand vernichtet.
Aber denk daran: Quantencomputer arbeiten normalerweise in einer Qubit-Basis mit spezifischen Regeln für die Darstellung fermionischer Systeme; sie arbeiten nicht inhärent in der Sprache fermionischer Operatoren. Um diese Lücke zu überbrücken, müssen wir diese fermionische Notation auf Pauli-Operatoren abbilden, die natürlicherweise Quantengates entsprechen.
Es gibt mehrere solche Transformationen für Fermionen. Eine gängige Wahl ist die Jordan-Wigner-Transformation. Die Bravyi-Kitaev- und Paritätsabbildungen sind neuere fermionische Kodierungen. Bosonische Operatoren können durch die Holstein-Primakoff-Transformation oder durch direkte Abbildung der Fock-Zustände auf eine Unterbasis der verfügbaren bosonischen Moden transformiert werden. Andere Kodierungen werden ebenfalls aktiv erforscht. Für jetzt werden wir uns nur auf die Jordan-Wigner-Transformation konzentrieren.
Die Jordan-Wigner-Transformation beinhaltet die Abbildung eines einzelnen Fermions auf viele Qubits. Warum können wir nicht einfach ein Qubit zuweisen, um jedes Elektron darzustellen? Das hängt mit der Unterscheidbarkeit identischer Fermionen zusammen. Qubits sind unterscheidbar und Elektronen nicht. Zum Beispiel können wir einzelne Qubits auf jedem Gerät leicht beschriften und identifizieren. Aber die Ununterscheidbarkeit von Elektronen bedeutet, dass wir sie überhaupt nicht beschriften können. Daher müssen wir die Operatoren tatsächlich gemäß besetzten Orbitalen beschriften, wie 1s, 2p, 2p usw., anstatt spezifischen Elektronen. Jedes Qubit spielt also ungefähr die Rolle eines Orbitals im Molekül, das entweder besetzt oder unbesetzt ist. Aber wie wir das tun, ist etwas komplizierter. Die Jordan-Wigner-Abbildung verfolgt die Antisymmetrie und stellt die korrekten Statistiken des gesamten fermionischen Systems sicher. Die Jordan-Wigner-Abbildung drückt fermionische Operatoren in Termen von Pauli-Operatoren mit diesen Beziehungen aus:
Die Jordan-Wigner-Abbildung ist konzeptionell einfach wegen der Eins-zu-Eins-Entsprechung zwischen Orbitalen und Qubits. Es gibt andere Abbildungen, die ein ähnliches Ziel erreichen, einschließlich der Paritätsabbildung. Die Berechnung der Parität eines Zustands erfordert die Berücksichtigung mehrerer Qubits. In der Paritätsabbildung (und einigen anderen) gilt die Interpretation, dass ein Qubit einem Orbital entspricht, nicht. Lass uns nun ein einfaches Beispiel durchgehen. Angenommen, wir möchten die Einzel-Qubit-Wechselwirkung berechnen. Wir beginnen damit, unsere Definitionen für die Erzeugungs- und Vernichtungsoperatoren einzusetzen.
Der Kommutator . Der endgültige Ausdruck wird also:
Wir haben also erfolgreich einen fermionischen Ausdruck in Pauli-Operatoren umgeschrieben, die unser Quantencomputer verstehen kann.
Lass uns kurz besprechen, wie wir die Jordan-Wigner-Abbildung in Qiskit implementieren würden. Es ist wichtig zu verstehen, wie diese Art von Transformationen funktioniert, aber es wäre unpraktisch, sie jedes Mal manuell auszuführen – besonders für große Systeme. Glücklicherweise macht Qiskit dies für uns mit der SparsePauliOp-Funktion einfach.
Auf einer hohen Ebene sind die Schritte:
- Verwende die
from_list-Funktion von SparsePauliOp, um einen Identitätsoperator zu erstellen, der der Größe des zu kartierenden Parameterraums entspricht. - Verwende gemäß der Definition der Erzeugungs- und Vernichtungsoperatoren, die zuvor gezeigt wurden, die
from_list-Funktion von SparsePauliOp, um -, -,-Pauli-Operatoren zu definieren. Dies wird die fermionischen Erzeugungs- und Vernichtungsoperatoren auf Qubit-Spin-Operatoren abbilden und die fermionische Besetzungszahl in die Rechenbasis von Qubits kodieren. - Erzeuge den gewünschten Hamiltonianer in der Pauli-Basis, indem du die obigen Operatoren auf die Orbitale von Interesse anwendest. Dies entspricht normalerweise der Erstellung einer Identitätsmatrix, die die Kern-(Nicht-Wechselwirkungs-)Orbitale darstellt, und dann dem Anwenden der Erzeugungs- und Vernichtungsoperatoren auf den aktiven Raum mit Gewichten, die den Besonderheiten des vorliegenden Problems entsprechen.
Nachdem wir das Jordan-Wigner-Abbildungsschema vollständig verstanden haben, lass uns ein komplizierteres Beispiel ansehen. Du erinnerst dich vielleicht an das Paper mit dem Titel „Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits" aus der vorherigen Lektion. Wir werden das Paper nicht nochmals im Detail durchgehen – wir werden uns nur auf die Jordan-Wigner-Abbildung konzentrieren, die verwendet wird, um den Hamiltonianer von Gitterpunkten mit Stellen für das Schwinger-Modell in Abwesenheit eines elektrischen Feldes auszudrücken.
Hier ist es viel schwieriger, genau zu bestimmen, was ein Qubit in diesem Modell darstellt, weil es nur die Sammlung von Qubits zusammen ist, die etwas Physisches ergibt, in diesem Fall ein Wellenpaket. Stattdessen kannst du die Qubits grob als diskretisierte Raumstücke betrachten. Hier ist das Gittervolumen, bei dem jedes Element (Einheitszelle) zwei Qubits umfasst. Die fermionischen Operatoren, die wir in der vorherigen Folie gesehen haben, beschreiben die Amplitude der Wellenfunktion an einem bestimmten Gitterplatz. Unser Hamiltonianer enthält also diese fermionischen Erzeugungs- und Vernichtungsoperatoren. Wir verwenden daher die Jordan-Wigner-Transformation, um diese Operatoren in die Pauli-Operatoren abzubilden.
wobei der Pauli-Operator und ist. Sobald wir einen Hamiltonianer in diesem Format haben, ist der schwierige Teil der Mapping-Phase vorbei, und er kann nun leicht in einen Circuit in Pauli-Operatoren geschrieben werden.
Fazit
Wir haben vier Beispiele diskutiert, wie spezifische Mapping-Techniken in letzter Zeit im Quantencomputing-Bereich eingesetzt wurden, angefangen vom Einfachsten bis hin zur Anwendung der Jordan-Wigner-Transformation auf Hadronendynamik. Vieles dieses Materials war sehr technisch, und wenn du es noch nicht gesehen hast, kann es sehr einschüchternd wirken. Aber es wird einfacher, je mehr Zeit du mit Üben verbringst – weshalb dieser Kurs „Quantum Computing in Practice" heißt. Das ist nichts, das jemand einfach aufnehmen und von Anfang an damit loslaufen kann – es erfordert etwas Studium, etwas Kopfkratzen und wahrscheinlich einige frustrierende Zeiten. Aber ich ermutige dich, bei diesem Unbehagen zu bleiben und die Fragen wirklich zu erforschen, die entstehen, wenn du weitermachst. Es ist der einzige Weg zu lernen.