Stichprobenbasierte Quantendiagonalisierung eines Chemie-Hamiltonoperators
Geschätzter Ressourcenverbrauch: weniger als eine Minute auf einem Heron-r2-Prozessor (HINWEIS: Dies ist nur eine Schätzung. Deine Laufzeit kann abweichen.)
Lernziele
Nach dem Durcharbeiten dieses Tutorials sollten die Nutzer/innen Folgendes verstehen:
- Wie man das SQD-Qiskit-Addon verwendet, um die Grundzustandsenergie eines molekularen Systems mithilfe von Bitstrings anzunähern, die von einer Quantenverarbeitungseinheit (QPU) abgetastet wurden.
- Wie man ffsim verwendet, um einen lokalen unitären Cluster-Jastrow-Ansatz (LUCJ) für die Quantenchemiesimulation zu konstruieren.
Voraussetzungen
Wir empfehlen, dass du dich mit den folgenden Themen vertraut machst, bevor du dieses Tutorial durcharbeitest:
- Quantenchemie und zweite Quantisierung
- Verwendung des Sampler-Primitives zur Abtastung von Quantenschaltkreisen
Hintergrund
In diesem Tutorial zeigen wir, wie man verrauschte Quantenstichproben nachbearbeitet, um den Grundzustand des Stickstoffmoleküls bei der Gleichgewichtsbindungslänge anzunähern, indem man das SQD-Qiskit-Addon verwendet, um den stichprobenbasierten Quantendiagonalisierungsalgorithmus (SQD) zu implementieren. Weitere Details zur Software sind in der entsprechenden Dokumentation zu finden, einschließlich eines einfachen Einstiegsbeispiels.
Dieses Tutorial richtet sich an Nutzer/innen, die mit der Quantenchemie vertraut sind: insbesondere mit der Bestimmung der Grundzustandsenergien eines Moleküls. Für eine detaillierte Beschreibung des Workflows wird auf den Kurs zum Quantendiagonalisierungsalgorithmus verwiesen.
SQD ist eine Technik zum Auffinden von Eigenwerten und Eigenvektoren von Quantenoperatoren, wie etwa einem Quanten-System-Hamiltonoperator, indem Quanten- und verteiltes klassisches Computing gemeinsam eingesetzt werden. Verteiltes klassisches Computing wird verwendet, um Stichproben zu verarbeiten, die von einem Quantenprozessor gewonnen wurden, und den Ziel-Hamiltonoperator in einem durch die Stichproben aufgespannten Unterraum zu projizieren und zu diagonalisieren. Ein SQD-basierter Workflow hat die folgenden Schritte:
- Wähle einen Schaltkreisansatz und wende ihn auf einem Quantencomputer auf einen Referenzzustand an (in diesem Fall den Hartree-Fock-Zustand).
- Taste Bitstrings aus dem resultierenden Quantenzustand ab.
- Führe das Verfahren der selbstkonsistenten Konfigurationswiederherstellung auf den Bitstrings aus, um die Grundzustandsnäherung zu erhalten.
Es ist bekannt, dass SQD gut funktioniert, wenn der Ziel-Eigenzustand dünn besetzt ist: Die Wellenfunktion wird in einer Menge von Basiszuständen dargestellt, deren Größe nicht exponentiell mit der Problemgröße wächst.
Quantenchemie
Der Hamiltonoperator eines molekularen Systems lässt sich schreiben als
wobei und komplexe Zahlen sind, die als Molekülintegrale bezeichnet werden und aus der Spezifikation des Moleküls mithilfe eines Computerprogramms berechnet werden können. In diesem Tutorial berechnen wir die Integrale mit dem Softwarepaket PySCF.
Für Details zur Herleitung des molekularen Hamiltonoperators konsultiere ein Lehrbuch zur Quantenchemie (zum Beispiel Modern Quantum Chemistry von Szabo und Ostlund). Für eine übergeordnete Erklärung, wie Quantenchemieprobleme auf Quantencomputer abgebildet werden, empfehlen wir die Vorlesung Mapping Problems to Qubits der Qiskit Global Summer School 2024.
Lokaler unitärer Cluster-Jastrow-Ansatz (LUCJ)
SQD benötigt einen Quantenschaltkreisansatz, aus dem Stichproben gezogen werden können. In diesem Tutorial verwenden wir den lokalen unitären Cluster-Jastrow-Ansatz (LUCJ) aufgrund seiner Kombination aus physikalischer Motivation und Hardware-Freundlichkeit. Wir verwenden ffsim, um den Ansatzschaltkreis zu konstruieren.
Der LUCJ-Ansatz passt sich an QPUs mit eingeschränkter Qubit-Konnektivität an. Die Spinorbitale werden so auf Qubits abgebildet, dass der Ansatz kein Routing mit SWAP Gates erfordert. IBM®-Hardware hat eine Heavy-Hex-Gitter-Qubit-Topologie, in diesem Fall können wir ein „Zickzack"-Muster verwenden, das unten dargestellt ist. In diesem Muster werden Orbitale mit demselben Spin auf Qubits mit einer Linientopologie abgebildet (rote und blaue Kreise), und eine Verbindung zwischen Orbitalen verschiedenen Spins besteht an jedem 4. räumlichen Orbital, wobei die Verbindung durch ein Ancilla-Qubit (lila Kreise) hergestellt wird.
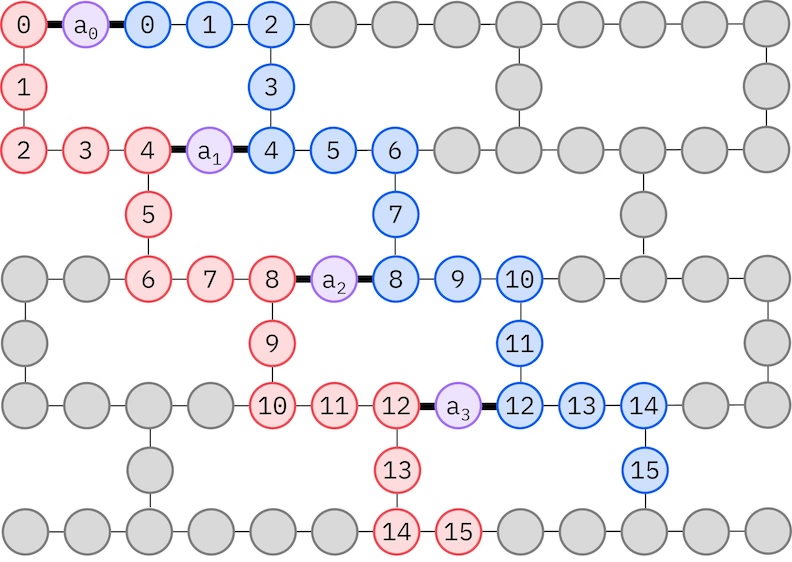
Selbstkonsistente Konfigurationswiederherstellung
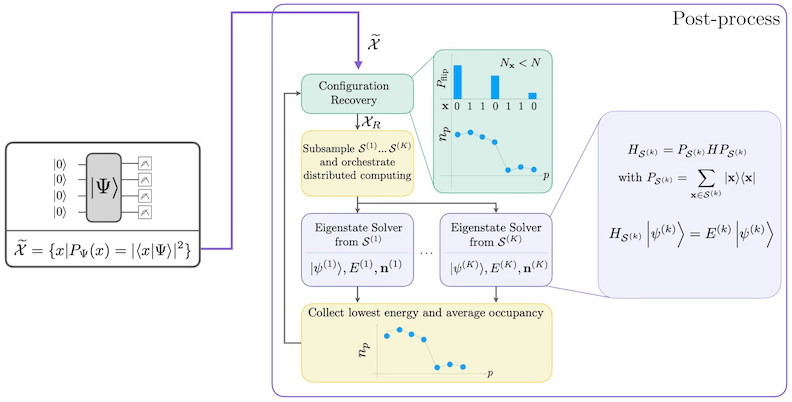
Das Verfahren der selbstkonsistenten Konfigurationswiederherstellung ist darauf ausgelegt, so viel Signal wie möglich aus verrauschten Quantenstichproben zu extrahieren. Da der molekulare Hamiltonoperator die Teilchenzahl und Spin-Z erhält, ist es sinnvoll, einen Schaltkreisansatz zu wählen, der diese Symmetrien ebenfalls erhält. Wenn er auf den Hartree-Fock-Zustand angewendet wird, hat der resultierende Zustand im rauschfreien Fall eine feste Teilchenzahl und Spin-Z. Daher sollte die Spin-- und Spin--Hälfte jedes Bitstrings, der aus diesem Zustand abgetastet wird, dasselbe Hamming-Gewicht wie im Hartree-Fock-Zustand haben. Aufgrund von Rauschen in aktuellen Quantenprozessoren verletzen einige gemessene Bitstrings diese Eigenschaft. Eine einfache Form der Nachselektion würde diese Bitstrings verwerfen, aber das ist verschwenderisch, da diese Bitstrings möglicherweise noch ein gewisses Signal enthalten. Das Verfahren der selbstkonsistenten Wiederherstellung versucht, einen Teil dieses Signals in der Nachbearbeitung wiederzugewinnen. Das Verfahren ist iterativ und benötigt als Eingabe eine Schätzung der mittleren Besetzungszahlen jedes Orbitals im Grundzustand, die zunächst aus den Rohstichproben berechnet wird. Das Verfahren wird in einer Schleife ausgeführt, und jede Iteration hat die folgenden Schritte:
- Für jeden Bitstring, der die angegebenen Symmetrien verletzt, werden seine Bits mit einem probabilistischen Verfahren umgekehrt, das darauf ausgelegt ist, den Bitstring näher an die aktuelle Schätzung der mittleren Orbitalbesetzungszahlen heranzubringen, um einen neuen Bitstring zu erhalten.
- Sammle alle alten und neuen Bitstrings, die die Symmetrien erfüllen, und nehme Teilmengen einer vorher festgelegten festen Größe als Teilstichproben.
- Projiziere für jede Teilmenge von Bitstrings den Hamiltonoperator in den durch die entsprechenden Basisvektoren aufgespannten Unterraum (siehe den vorherigen Abschnitt für eine Beschreibung dieser Basisvektoren) und berechne auf einem klassischen Computer eine Grundzustandsnäherung des projizierten Hamiltonoperators.
- Aktualisiere die Schätzung der mittleren Orbitalbesetzungszahlen mit der Grundzustandsnäherung mit der niedrigsten Energie.
SQD-Workflow-Diagramm
Der SQD-Workflow ist im folgenden Diagramm dargestellt:
Anforderungen
Bevor du dieses Tutorial beginnst, stelle sicher, dass du Folgendes installiert hast:
- Qiskit SDK v1.0 oder höher, mit Unterstützung für Visualisierung
- Qiskit Runtime v0.22 oder höher (
pip install qiskit-ibm-runtime) - SQD-Qiskit-Addon v0.11 oder höher (
pip install qiskit-addon-sqd) - ffsim v0.0.75 oder höher (
pip install ffsim)
Einrichtung
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Kleinskaliges Simulatorbeispiel
In diesem Tutorial werden wir eine Näherung an den Grundzustand eines Stickstoffmoleküls in der Nähe seines Gleichgewichtsbindungsabstands finden. Wir verwenden zunächst einen kleinen STO-6G-Basissatz, damit wir das Experiment simulieren und sicherstellen können, dass es funktioniert.
Schritt 1: Klassische Eingaben auf ein Quantenproblem abbilden
Zuerst geben wir das Molekül und seine Eigenschaften an.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Bevor wir den LUCJ-Ansatzschaltkreis konstruieren, führen wir zunächst eine CCSD-Berechnung in der folgenden Codezelle durch. Die - und -Amplituden aus dieser Berechnung werden verwendet, um die Parameter des Ansatzes zu initialisieren.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Nun verwenden wir ffsim, um den Ansatzschaltkreis zu erstellen. Da unser Molekül einen geschlossenschaligen Hartree-Fock-Zustand hat, verwenden wir die spinbalancierte Variante des UCJ-Ansatzes, UCJOpSpinBalanced. Wir setzen optimize=True in der from_t_amplitudes-Methode, um die „komprimierte" Doppelfaktorisierung der -Amplituden zu aktivieren (siehe The local unitary cluster Jastrow (LUCJ) ansatz in der ffsim-Dokumentation für Details).
Da der LUCJ-Ansatz sich an die verfügbare Konnektivität der QPU anpasst, müssen wir das QPU-Backend initialisieren, bevor wir den Ansatz erstellen. Zunächst erstellen wir ein generisches Backend mit einer Heavy-Hex-Kopplungskarte und einem Gate-Satz, in den sich der LUCJ-Ansatz natürlich zerlegen lässt. Dann verwenden wir ffsim.qiskit.generate_lucj_pass_manager, um einen Pass-Manager zu erstellen, der spezialisiert ist auf die Transpilierung des LUCJ-Ansatzes auf das gegebene Backend gemäß dem im Hintergrundabschnitt zum LUCJ-Ansatz beschriebenen „Zickzack"-Layout. Diese Funktion verwendet eine Bewertungsheuristik, um die mit dem ausgewählten Layout verbundenen Fehler zu minimieren, was wichtig ist, wenn dein Backend eine echte QPU oder ein Simulator mit einem Rauschmodell ist. Neben der Rückgabe des Pass-Managers gibt diese Funktion auch die Alpha-Beta-Kopplungspaare zurück, die auf der Hardware implementiert werden können. Wenn nicht alle Paare implementiert werden können, gibt sie eine Warnung aus.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Schritt 2: Für die Quantenhardware-Ausführung optimieren
Als nächstes optimieren wir den Circuit für eine Zielhardware. Typischerweise beinhaltet dieser Schritt die Initialisierung des Hardware-Backends und eines Pass-Managers für dieses Backend. Da der LUCJ-Ansatz jedoch an die Hardware-Konnektivität angepasst ist, haben wir diese Schritte bereits im vorherigen Schritt durchgeführt. Alles, was noch zu tun ist, ist den Pass-Manager auf dem Circuit auszuführen, um ihn in einen ISA-Circuit zu transpilieren, der direkt auf der QPU ausgeführt werden kann.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Schritt 3: Mit Qiskit-Primitives ausführen
Nachdem wir den Circuit für die Hardware-Ausführung optimiert haben, sind wir bereit, ihn auf der Zielhardware auszuführen und Stichproben für die Grundzustandsenergieabschätzung zu sammeln. Da wir nur einen Circuit haben, werden wir den Job-Ausführungsmodus von Qiskit Runtime verwenden und unseren Circuit ausführen.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Schritt 4: Nachbearbeiten und Ergebnis im gewünschten klassischen Format zurückgeben
Eine nützliche Kenngröße zur Beurteilung der Qualität der QPU-Ausgabe ist die Anzahl der zurückgegebenen gültigen Konfigurationen. Eine gültige Konfiguration hat die korrekte Teilchenzahl und Spin-Z, was bedeutet, dass die rechte Hälfte des Bitstrings das Hamming-Gewicht gleich der Anzahl der Spin-up-Elektronen hat und die linke Hälfte das Hamming-Gewicht gleich der Anzahl der Spin-down-Elektronen. Die folgende Zelle berechnet den Anteil der abgetasteten Konfigurationen, die gültig sind.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Alle Bitstrings sind gültig, weil wir den Circuit auf einem rauschfreien Simulator abtasten. Bei der Ausführung auf einer verrauschten QPU ist der Anteil kleiner als eins, aber hoffentlich größer als der Anteil, den man erwarten würde, wenn die Bitstrings gleichmäßig zufällig abgetastet würden, was in der folgenden Zelle berechnet wird.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Nun schätzen wir die Grundzustandsenergie des Hamiltonoperators mithilfe der Funktion diagonalize_fermionic_hamiltonian. Diese Funktion führt das Verfahren der selbstkonsistenten Konfigurationswiederherstellung durch, um die verrauschten Quantenstichproben iterativ zu verfeinern und die Energieschätzung zu verbessern. Wir übergeben eine Callback-Funktion, damit wir die Zwischenergebnisse für spätere Analysen speichern können. Erklärungen zu den Argumenten von diagonalize_fermionic_hamiltonian findest du in der API-Dokumentation.
Hier verwenden wir das Argument initial_occupancies für diagonalize_fermionic_hamiltonian, um die Hartree-Fock-Konfiguration als anfängliche Schätzung für die Orbitalbesetzungszahlen im Grundzustand anzugeben. Dieser Ansatz ist sinnvoll für Systeme, bei denen der Grundzustand signifikante Unterstützung auf der Hartree-Fock-Konfiguration hat, aber er könnte in anderen Situationen ungeeignet sein, obwohl fortgeschrittenere Rechenmethoden in diesen Fällen bessere Anfangsschätzungen liefern könnten. Die Angabe von initial_occupancies ermöglicht es außerdem, die Konfigurationswiederherstellung auch dann auszuführen, wenn keine gültigen Konfigurationen abgetastet wurden, was beim Abtasten eines großen Circuits auf einer verrauschten QPU der Fall sein kann. Ohne dieses Argument würde die Konfigurationswiederherstellung fehlschlagen und einen Fehler auslösen, wenn keine gültigen Konfigurationen bereitgestellt wurden.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Ergebnisse visualisieren
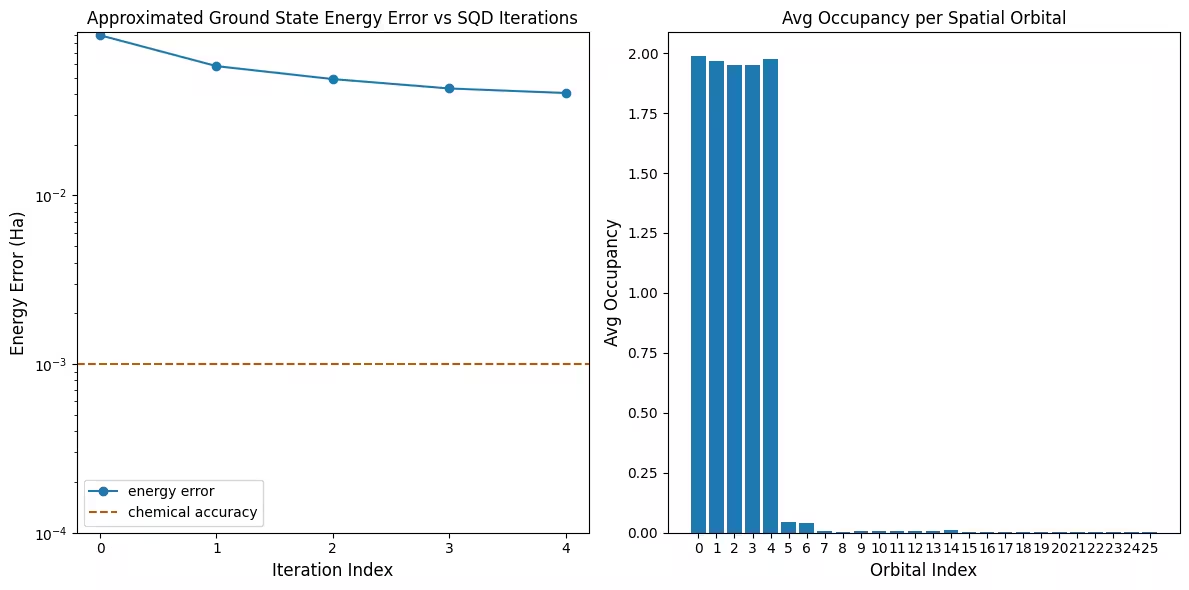
Der erste Plot zeigt, dass wir in dieser Simulation bereits nach der ersten Iteration innerhalb von 1 mH der genauen Antwort liegen (chemische Genauigkeit wird typischerweise als 1 kcal/mol 1.6 mH akzeptiert). Dies ist jedoch ein kleines System, und da die Stichproben rauschfrei sind, ist keine Konfigurationswiederherstellung notwendig. Bei einem größeren System, das auf einer verrauschten QPU ausgeführt wird, könnten mehrere Iterationen der Konfigurationswiederherstellung erforderlich sein, und die endgültige Genauigkeit könnte schlechter sein. Im Allgemeinen kann die Energie verbessert werden, indem mehr Iterationen der Konfigurationswiederherstellung zugelassen werden oder indem die Anzahl der Stichproben pro Batch erhöht wird.
Der zweite Plot zeigt die durchschnittliche Besetzungszahl jedes räumlichen Orbitals nach der letzten Iteration. Man sieht, dass sowohl die Spin-up- als auch die Spin-down-Elektronen in unseren Lösungen mit hoher Wahrscheinlichkeit die ersten fünf Orbitale besetzen.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Großskaliges Hardware-Beispiel
Nun führen wir ein größeres Beispiel auf echter Quantenhardware aus. Hier leiten wir einen aktiven Raum für das Stickstoffmolekül aus dem cc-pVDZ-Basissatz ab.
Schritte 1–4
Hier fassen wir alle Schritte zu einem einzigen Workflow in größerem Maßstab zusammen, der dann auf echter Quantenhardware ausgeführt wird.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Nächste Schritte
Wenn du diese Arbeit interessant findest, könnten dich die folgenden Materialien interessieren:
- Stichprobenbasierte Krylov-Quantendiagonalisierung eines fermionischen Gittermodells – ein verwandtes Tutorial, das Zeitentwicklungsschaltkreise anstelle eines variationellen Ansatzes verwendet
- SQD-Chemie-Workflows mit Dice-Solver skalieren – eine Seite, die zeigt, wie man die effizientere Dice-Software für die Diagonalisierung verwendet
- SQD-Addon-API-Dokumentation – Referenz für die Funktion
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer – das Paper, auf dem dieses Tutorial basiert