Variational Quantum Eigensolver (VQE)
Für dieses Modul benötigst du eine funktionierende Python-Umgebung sowie die neuesten Versionen der folgenden Pakete:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Informationen zur Einrichtung und Installation dieser Pakete findest du im Qiskit installieren-Leitfaden. Um Jobs auf echten Quantencomputern auszuführen, musst du ein IBM Cloud-Konto einrichten – die nötigen Schritte sind im Leitfaden IBM Cloud-Konto einrichten beschrieben.
Dieses Modul wurde getestet und hat dabei ungefähr 8 Minuten QPU-Zeit verbraucht. Dies ist ein Schätzwert – dein tatsächlicher Verbrauch kann abweichen.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Einführung
Seit der Entwicklung des quantenmechanischen Modells im frühen 20. Jahrhundert wissen Wissenschaftler, dass Elektronen keine festen Bahnen um den Atomkern beschreiben, sondern sich in Wahrscheinlichkeitsbereichen aufhalten, die als Orbitale bezeichnet werden. Diese Orbitale entsprechen bestimmten, diskreten Energieniveaus, die Elektronen besetzen können. Elektronen befinden sich natürlicherweise auf den niedrigsten verfügbaren Energieniveaus, dem sogenannten Grundzustand. Nimmt ein Elektron jedoch genügend Energie auf, kann es auf ein höheres Energieniveau springen und einen angeregten Zustand einnehmen. Dieser angeregte Zustand ist vorübergehend, und das Elektron kehrt schließlich auf ein niedrigeres Energieniveau zurück, wobei es die aufgenommene Energie – häufig in Form von Licht – wieder abgibt. Dieser grundlegende Prozess der Energieaufnahme und -abgabe ist entscheidend für das Verständnis, wie Atome miteinander wechselwirken und Bindungen eingehen.
Wenn Atome zusammenkommen und Moleküle bilden, verbinden sich ihre Atomorbitale zu Molekülorbitalen. Die Anordnung und die Energieniveaus der Elektronen in diesen Molekülorbitalen bestimmen die Eigenschaften des entstehenden Moleküls sowie die Stärke der chemischen Bindungen. Bei der Bildung eines Wasserstoffmoleküls () aus zwei einzelnen Wasserstoffatomen zum Beispiel besetzt das Elektron jedes Atoms zunächst ein Atomorbital. Wenn die Atome sich annähern, überlappen sich diese Atomorbitale und bilden neue Molekülorbitale – eines mit niedrigerer Energie (ein bindendes Orbital) und eines mit höherer Energie (ein antibindendes Orbital). Die beiden Elektronen, je eines von jedem Wasserstoffatom, besetzen bevorzugt das energieärmere bindende Orbital, was zur Ausbildung einer stabilen kovalenten Bindung führt, die das -Molekül zusammenhält. Der Energieunterschied zwischen den getrennten Atomen und dem gebildeten Molekül – insbesondere die Energie der Elektronen in den Molekülorbitalen – bestimmt die Stabilität und die Eigenschaften der Bindung.
In den folgenden Abschnitten werden wir diesen Prozess der Molekülbildung am Beispiel des -Moleküls untersuchen. Wir verwenden einen echten Quantencomputer in Kombination mit klassischen Optimierungsverfahren, um die Energie dieses einfachen, aber grundlegenden Prozesses zu berechnen. Dieses Experiment bietet eine praktische Demonstration, wie Quantencomputing auf Probleme der computergestützten Chemie angewendet werden kann und Einblicke in die Rolle der Elektronenenergie liefert.
VQE – Ein variationeller Quantenalgorithmus für Eigenwertprobleme
Näherungsverfahren in der Chemie – Variationsprinzip und Basissatz
Erwin Schrödingers Beiträge zur Quantenmechanik beschränken sich nicht auf die Einführung eines neuen Elektronenmodells; grundlegend hat er die Wellenmechanik begründet, indem er die berühmte zeitabhängige Schrödinger-Gleichung entwickelte:
Hierbei ist der Hamiltonoperator, der die Gesamtenergie des Systems repräsentiert, und ist die Wellenfunktion, die alle Informationen über den Quantenzustand des Systems enthält. (Hinweis: ist die totale Zeitableitung, und der Energieeigenwert erscheint hier nicht explizit.)
In vielen praktischen Anwendungen – etwa bei der Bestimmung der erlaubten Energieniveaus von Atomen und Molekülen – verwenden wir stattdessen die zeitunabhängige Schrödinger-Gleichung (Energieeigenwertgleichung), die aus der zeitabhängigen Form abgeleitet wird, indem man einen stationären Zustand voraussetzt. Ein stationärer Zustand ist ein Quantenzustand, bei dem sich die Wahrscheinlichkeitsdichte, ein Teilchen an einem bestimmten Ort zu finden, zeitlich nicht ändert.
In dieser Form steht für den Energieeigenwert, der dem Quantenzustand entspricht. Der Hamiltonoperator umfasst verschiedene Energiebeiträge, wie die kinetische Energie von Elektronen und Atomkernen, die Anziehungskräfte zwischen Elektronen und Atomkernen sowie die Abstoßungskräfte zwischen Elektronen.
Das Lösen der Energieeigenwertgleichung ermöglicht uns die Berechnung der quantisierten Energieniveaus von Atom- und Molekülsystemen. Bei Molekülen ist eine exakte Lösung jedoch schwierig, da die Wellenfunktion , die die räumliche Verteilung der Elektronen beschreibt, komplex und hochdimensional ist.
Daher verwenden Wissenschaftler Näherungsverfahren, um praktikable und genaue Lösungen zu erhalten. In dieser Arbeit konzentrieren wir uns auf zwei zentrale Methoden:
-
Variationsprinzip
Diese Methode nähert die Wellenfunktion an und passt sie so an, dass sie der Zielenergie – in der Regel der Grundzustandsenergie des Systems – so nahe wie möglich kommt. Der Kerngedanke hinter dem Variationsprinzip ist einfach:
- Wenn wir eine Wellenfunktion schätzen (eine „Testfunktion"), ist die daraus berechnete Energie stets gleich oder größer als die Grundzustandsenergie () des Systems.
- Durch Anpassen der Parameter in der Testfunktion, , lässt sich die Grundzustandsenergie immer besser annähern.
- Die Genauigkeit hängt stark von der Wahl der Testwellenfunktion ab. Eine schlecht gewählte Testfunktion kann zu einer Energieschätzung führen, die weit von der tatsächlichen Energie entfernt ist.
-
Basissatznäherung
Die zweite Näherungsmethode kommt bei der Konstruktion der Wellenfunktion zum Einsatz – dem Basissatz-Ansatz. In der Quantenchemie ist es nahezu unmöglich, die Schrödinger-Gleichung für Moleküle exakt zu lösen. Stattdessen nähern wir die komplexe Mehrelektronenwellenfunktion an, indem wir sie aus einfacheren, vordefinierten mathematischen Funktionen aufbauen. Ein Basissatz ist im Wesentlichen eine Sammlung solcher bekannten mathematischen Funktionen, die typischerweise auf den Atomen des Moleküls zentriert sind und als Bausteine dienen, um die Form und das Verhalten der Elektronen im System darzustellen. Stell es dir vor wie den Versuch, eine detaillierte Skulptur nur aus handelsüblichen LEGO-Steinen nachzubauen – je mehr Arten und Größen von Steinen du hast (je größer der Basissatz), desto genauer kannst du die ursprüngliche Form annähern.
Diese Basisfunktionen sind häufig von den analytischen Lösungen einfacher Systeme wie dem Wasserstoffatom inspiriert und nehmen Formen wie Gauß- oder Slater-Funktionen an, obwohl auch sie nur Näherungen sind. Statt mit den theoretisch „exakten", aber unlösbaren vollständigen Molekülorbitalen zu arbeiten, drücken wir diese als Linearkombination (eine gewichtete Summe) dieser Basisfunktionen aus. Diese Methode wird als LCAO-Ansatz (Linear Combination of Atomic Orbitals) bezeichnet, wenn die Basisfunktionen Atomorbitalen ähneln. Durch Optimierung der Koeffizienten in dieser Linearkombination lässt sich die bestmögliche angenäherte Wellenfunktion und Energie innerhalb der Grenzen des gewählten Basissatzes finden.
- Je mehr Funktionen der Basissatz enthält, desto besser die Näherung – allerdings auf Kosten eines höheren Rechenaufwands.
- Ein kleiner Basissatz liefert eine grobe Schätzung, während ein großer Basissatz präzisere Ergebnisse auf Kosten von mehr Rechenressourcen liefert.
Zusammenfassend lässt sich sagen: Um Berechnungen handhabbar zu machen und den Rechenaufwand zu reduzieren, verwenden wir das Variationsprinzip zur Annäherung der Wellenfunktion, was die Rechenkomplexität verringert und eine iterative Optimierung zur Energieminimierung ermöglicht. Der Basissatz-Ansatz vereinfacht Berechnungen, indem Atomorbitale als Kombination vordefinierter Funktionen dargestellt werden, anstatt direkt eine kontinuierliche Wellenfunktion zu lösen.
Verständnisfragen
Betrachte die Testwellenfunktion , wobei eine Normierungskonstante und ein einstellbarer Parameter ist.
(a) Normiere die Testwellenfunktion, indem du so bestimmst, dass .
Lösung
Normierung der gegebenen Testwellenfunktion:
Verwende das Gaußsche Integral:
Setze und erhalte:
(b) Berechne den Erwartungswert des Hamiltonoperators gemäß wobei dem Potential eines harmonischen Oszillators entspricht.
Lösung
Der Hamiltonoperator für einen harmonischen Oszillator lautet:
Erwartungswert der kinetischen Energie
Berechnung der zweiten Ableitung:
Damit gilt:
Unter Verwendung der Standardergebnisse für Gaußsche Integrale:
Erwartungswert der potenziellen Energie
Mit:
erhalten wir:
Gesamtenergie-Erwartungswert
(c) Verwende das Variationsprinzip, um das optimale durch Minimierung von zu finden.
Lösung
Optimierung von für minimale Energie
Ableitung bilden
Auflösen:
Einsetzen von in :
was genau der Grundzustandsenergie des quantenmechanischen harmonischen Oszillators entspricht.
VQE (Variational Quantum Eigensolver)
Der Variational Quantum Eigensolver (VQE) ist die zentrale Methode, die wir zur Untersuchung des Prozesses einsetzen werden. Hier schauen wir uns an, was VQE ist und wie es funktioniert. Doch zuerst halten wir kurz inne und betrachten eine besonders wichtige Frage.
Verständnisfragen
Wenn wir bereits so viele Strategien für Chemieprobleme haben – warum brauchen wir dann überhaupt einen Quantencomputer? Und wozu dient die Kombination aus Quanten- und Klassischem Computer?
Antwort
Quantencomputing hat das Potenzial, die Chemie zu revolutionieren, indem es Probleme angeht, mit denen klassische Computer aufgrund der exponentiellen Skalierung von Quantenzuständen zu kämpfen haben. Richard Feynman bemerkte treffend, dass zur Simulation der Natur auch die Berechnungen quantenmechanischer Natur sein müssen [Ref. 1].
Die Simulation von Koffein mit dem einfachsten Basissatz (STO-3G) würde beispielsweise Bits erfordern – weit mehr als die Gesamtzahl der Sterne im sichtbaren Universum () [Ref. 2]. Ein Quantencomputer kann die Elektronen-Orbitale von Koffein mit 160 Qubits beschreiben.
Quantencomputer verarbeiten Quantenwechselwirkungen auf natürliche Weise mithilfe von Superposition und Verschränkung, was einen vielversprechenden Weg für präzise Molekülsimulationen eröffnet. Darüber hinaus lassen sich die Vorteile beider Systeme kombinieren: Quantencomputer für die Elektronensimulation und klassische Computer für Datenvor- und -nachverarbeitung, Algorithmussteuerung, Optimierung und mehr. Dies soll Materialentdeckung, Wirkstoffdesign und Reaktionsvorhersagen vorantreiben und kostspielige Experimente nach dem Versuch-und-Irrtum-Prinzip reduzieren. [Ref. 3][Ref. 4]
Wenn du verstehen möchtest, warum Quantencomputer für Chemieprobleme benötigt werden und warum man Quanten- und klassische Rechenressourcen kombiniert, lies die folgenden Artikel:
Kehren wir nun zu VQE zurück.
VQE kombiniert die Stärke von Quantencomputern mit klassischen Computern und nutzt dabei grundlegend Variationsprinzipien, um die Grundzustandsenergie des Systems zu bestimmen. Um VQE zu verstehen, zerlegen wir es in drei Teile:
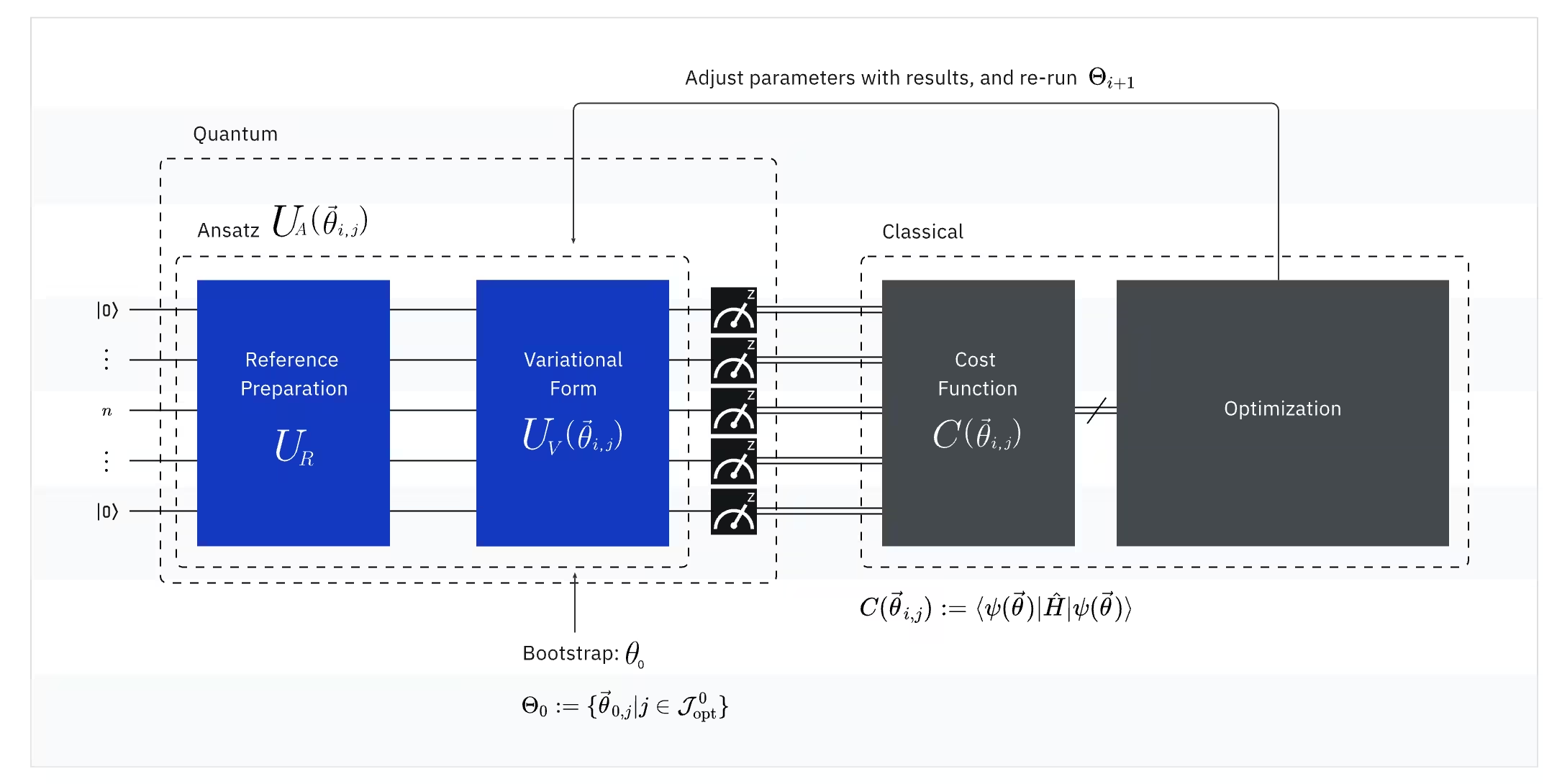
(Quanten-) Observable: Der molekulare Hamiltonoperator (Energie eines Moleküls)
Im VQE ist der molekulare bzw. atomare Hamiltonoperator eine Observable, das heißt, wir können seinen Wert durch ein Experiment messen. Unser Ziel ist es, die niedrigstmögliche Energie (die Grundzustandsenergie) des Moleküls zu finden. Dazu verwenden wir einen Testzustand, der durch einen parametrisierten Quantenschaltkreis (Ansatz) erzeugt wird. Wir messen die Observable und optimieren den Quantenzustand, bis wir die niedrigstmögliche Energie erreichen.
Der für den molekularen Hamiltonoperator verwendete Basissatz bestimmt die Anzahl der benötigten Qubits und beeinflusst direkt die Genauigkeit des VQE. Die Wahl des richtigen Basissatzes ist entscheidend für die Balance zwischen Effizienz und Präzision. Um Berechnungen zu vereinfachen, ohne den Basissatz zu ändern, können wir Strategien wie die Ausnutzung von Symmetrien und die Reduktion des aktiven Raums einsetzen. Viele Moleküle besitzen symmetrische Formen (wie ein Schmetterling oder eine Schneeflocke), was bedeutet, dass sich einige Teile gleich verhalten. Anstatt alles separat zu berechnen, können wir uns auf einzigartige Teile konzentrieren und damit Quantenressourcen sparen – das ist das Ausnutzen von Symmetrie. Bei der Reduktion des aktiven Raums berücksichtigen wir nur die wichtigen Orbitale, da nicht alle Elektronen die Molekülenergie wesentlich beeinflussen. Elektronen in Kernnähe bleiben weitgehend unverändert, während andere die Bindung beeinflussen. Durch diese Methoden lässt sich VQE effizienter gestalten, ohne an Genauigkeit einzubüßen.
Sobald wir einen molekularen Hamiltonoperator mit dem richtigen Basissatz und den oben genannten Strategien erhalten haben, müssen wir diesen Hamiltonoperator in eine Form transformieren, die für Quantencomputer geeignet ist. Die Abbildung von Problemen auf Pauli-Operatoren kann recht kompliziert sein. Das gilt insbesondere in der Quantenchemie, die mit ununterscheidbaren Teilchen (Elektronen) arbeitet, während Qubits unterscheidbar sind. Auf die Details der Abbildungen gehen wir hier nicht ein, verweisen aber auf folgende Ressourcen: Eine allgemeine Diskussion zur Abbildung eines Problems auf Quantenoperatoren findet sich in Quantum computing in practice. Eine ausführlichere Diskussion zur Abbildung chemischer Probleme auf Quantenoperatoren gibt es in Quantum chemistry with VQE.
Für dieses Modul stellen wir dir die geeigneten (Ein-Qubit-) Hamiltonoperatoren für und zur Verfügung, damit wir uns auf die Nutzung des Quantencomputers konzentrieren können. Diese Ein-Qubit-Hamiltonoperatoren wurden mithilfe des STO-6G-Basissatzes und der Jordan-Wigner-Abbildung erstellt, der direktesten Abbildung mit der einfachsten physikalischen Interpretation, da sie die Besetzung eines Spin-Orbitals auf die Besetzung eines Qubits abbildet. Außerdem verwendeten wir eine Qubit-Reduktionsmethode mithilfe einer Symmetrie des Hamiltonoperators, die Muster im Verhalten der Spin-Besetzungen nutzt, um die Anzahl der Qubits zu reduzieren. Für das -Molekül nehmen wir einen Abstand von 0.735 zwischen den beiden Wasserstoffatomen an.
(Quanten-) Ansatz: Die Testwellenfunktion (Wie man einen trivialen Quantenzustand mit einem Quantenschaltkreis aufbaut)
Beim VQE besteht der Ansatz (Plural: Ansätze) aus zwei wesentlichen Komponenten. Die erste ist die Vorbereitung des Anfangszustands, die den Zustand des Qubits durch Anwendung von Quantengattern ohne variationelle Parameter einstellt. Die zweite Komponente ist der parametrisierte Quantenschaltkreis – ein spezieller Quantenschaltkreis mit einstellbaren Parametern, ähnlich den Reglern an einem Radio. Diese Parameter werden im letzten Teil – dem klassischen Optimierer – verwendet, um den bestmöglichen Grundzustand zu erreichen.
Im Abschnitt zum Variationsprinzip haben wir gelernt, dass die Qualität des Testzustands die Qualität der Ergebnisse des Variationsalgorithmus beeinflusst. Das bedeutet, dass die Wahl eines guten Ansatzes bei VQE entscheidend ist. Auch dies ist ein umfangreiches und komplexes Thema. Wir werden die verschiedenen Typen von Ansätzen und ihre Herkunft hier nicht behandeln. Wenn du mehr über parametrisierte Quantenschaltkreise und Ansätze erfahren möchtest, empfehlen wir die Lektion Ansatz und Variationsform aus dem Kurs zum Design variationeller Algorithmen, die ausführliche Erklärungen und Beispiele für Ansätze bietet.
Da wir in diesem Modul einen Ein-Qubit-Hamiltonoperator verwenden, benötigen wir einen Ein-Qubit-parametrisierten Quantenschaltkreis als Ansatz. Im folgenden Abschnitt werden wir drei Typen von Ein-Qubit-Ansätzen kennenlernen, sie vergleichen und wichtige Überlegungen bei der Auswahl eines Ansatzes besprechen.
(Klassischer) Optimierer: Feinabstimmung des Quantenschaltkreises
Sobald der Quantencomputer die Energie der Observable aus dem Ansatz gemessen hat, werden die Parameter des Ansatzes und der Energiewert zur Abstimmung an den klassischen Optimierer übermittelt. Dieser Optimierungsprozess wird auf einem klassischen Computer durchgeführt, typischerweise unter Verwendung von wissenschaftlichen Allzweckpaketen wie SciPy.
Der klassische Optimierer behandelt die gemessene Energie als Kostenfunktion. Bei Optimierungsproblemen ist eine Kostenfunktion (auch Zielfunktion genannt) eine mathematische Funktion, die misst, wie „gut" eine bestimmte Lösung ist. Das Ziel des Optimierers ist es, den Parametersatz zu finden, der diese Kostenfunktion minimiert. Im Kontext der Suche nach der Grundzustandsenergie eines Moleküls dient die Energie selbst als Kostenfunktion – wir wollen die Parameter für unseren Quantenschaltkreis (unsere „Lösung") finden, die die niedrigstmögliche Energie ergeben. Der klassische Optimierer verwendet diesen gemessenen Energiewert (die Kosten) und bestimmt den nächsten Satz optimierter Parameter für den Quantenansatz. Diese aktualisierten Parameter werden dann zurück an den Quantenschaltkreis gesendet, und der Prozess wiederholt sich. Mit jeder Iteration passt der klassische Optimierer die Parameter an, um die Energie zu senken (die Kostenfunktion zu minimieren), bis ein vordefiniertes Konvergenzkriterium erfüllt ist – im Idealfall so, dass die niedrigstmögliche Energie (der Grundzustand des Moleküls bei diesem Bindungsabstand und Basissatz) gefunden wird.
Wissenschaftliche Pakete wie SciPy bieten viele Optimierungsstrategien. Mehr dazu findest du in der Lektion Optimierungsschleifen des Kurses Variational algorithm design. Hier verwenden wir COBYLA (Constrained Optimization BY Linear Approximations), einen Optimierungsalgorithmus, der für komplizierte Energielandschaften geeignet ist. COBYLA versucht insbesondere nicht, einen Gradienten der untersuchten Funktion zu berechnen; man nennt dies einen gradientenfreien Optimierer. Stell dir vor, du versuchst mit geschlossenen Augen den höchsten Gipfel in einem Gebirge zu finden. Da du die gesamte Landschaft nicht sehen kannst, machst du kleine Schritte in verschiedene Richtungen und prüfst dabei, ob es bergauf oder bergab geht. COBYLA funktioniert ähnlich – es bewegt sich durch den Parameterraum, testet verschiedene Werte und verbessert das Ergebnis schrittweise, bis es das beste findet.
Jetzt bist du bereit, eine VQE-Berechnung durchzuführen. Beantworte dazu die folgende Verständnisfrage, die den Gesamtprozess noch einmal zusammenfasst.
Verständnisfragen
Fülle die Lücken mit den richtigen Begriffen aus, um die Zusammenfassung des VQE-Prozesses zu vervollständigen, und klicke anschließend, um deine Antworten zu überprüfen.
VQE ist ein variationeller Quantenalgorithmus, der die Stärke von (1) ________ und klassischem Computing kombiniert und dazu dient, die (2) __________ eines Moleküls zu bestimmen. Der Prozess beginnt mit der Definition des (3) __________, der die Gesamtenergie des Systems repräsentiert und als Observable in Quantenmessungen dient. Als nächstes bereiten wir einen (4) __________ vor – einen Quantenschaltkreis mit einstellbaren Parametern, der die Testwellenfunktion des Moleküls darstellt. Diese Parameter werden mit einem (5) __________ optimiert, einem klassischen Algorithmus, der die Parameter iterativ anpasst, um die gemessene Energie zu minimieren. In der obigen Diskussion haben wir den (6) __________-Optimierer verwendet, der die Ansatz-Parameter ohne Gradientenberechnungen verfeinert. Der Prozess wird fortgesetzt, bis wir (7) __________ erreichen, das heißt, wir haben die niedrigstmögliche Energie des Moleküls gefunden.
Begriffe:
- klassischer Optimierer
- Grundzustandsenergie
- hardwareeffizient
- Ansatz
- molekularer Hamiltonoperator
- COBYLA
- Quantencomputing
- Konvergenz
Antwort
1 → Quantencomputing
2 → Grundzustandsenergie
3 → molekularer Hamiltonoperator
4 → Ansatz
5 → klassischer Optimierer
6 → COBYLA
7 → Konvergenz
Grundzustandsenergie eines Wasserstoffatoms mit VQE berechnen
Jetzt wenden wir das Gelernte an und berechnen die Grundzustandsenergie eines Wasserstoffatoms. Im gesamten Modul verwenden wir ein Framework für Quantencomputing namens „Qiskit Patterns", das Workflows in folgende Schritte unterteilt:
- Schritt 1: Klassische Eingaben auf ein Quantenproblem abbilden
- Schritt 2: Problem für die Quantenausführung optimieren
- Schritt 3: Mit Qiskit Runtime Primitives ausführen
- Schritt 4: Nachverarbeitung und klassische Analyse

Wir werden diese Schritte im Allgemeinen befolgen.
Beginnen wir damit, einige notwendige Pakete zu laden, darunter die Qiskit Runtime Primitives. Außerdem wählen wir den am wenigsten ausgelasteten verfügbaren Quantencomputer aus.
Im folgenden Code findest du Anweisungen zum Speichern deiner Zugangsdaten bei der ersten Verwendung. Stelle sicher, dass du diese Informationen nach dem Speichern in deiner Umgebung aus dem Notebook entfernst, damit deine Zugangsdaten nicht versehentlich weitergegeben werden, wenn du das Notebook teilst. Weitere Hinweise findest du unter IBM Cloud-Konto einrichten und Dienst in einer nicht vertrauenswürdigen Umgebung initialisieren.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Die folgende Zelle ermöglicht es dir, während des gesamten Notebooks zwischen dem Simulator und echter Hardware zu wechseln. Wir empfehlen, sie jetzt auszuführen:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Schritt 1: Das Problem auf Quantenschaltkreise und Operatoren abbilden
Wir beginnen unsere VQE-Berechnung, indem wir den Hamiltonoperator für das Wasserstoffmolekül () bei einem bestimmten Bindungsabstand definieren. Dieser Hamiltonoperator stellt die Gesamtenergie des Systems in Form von Qubit-Operatoren dar und wurde aus dem molekularen System mithilfe eines Standardverfahrens erzeugt und abgebildet: 1) Verwendung des STO-6G-Basissatzes (eine spezifische Sammlung mathematischer Funktionen zur Näherung der Elektronen-Orbitale), 2) Anwendung des Jordan-Wigner-Mappings (eine Technik zur Übersetzung fermionischer Operatoren, die Elektronen beschreiben, in Qubit-Operatoren) und 3) Qubit-Reduktion durch Ausnutzung der Parität des Hamiltonoperators, um das Problem zu vereinfachen.
Wie bereits erläutert, hängen die berechneten Grundzustandsenergien stark von der Wahl des Basissatzes und der molekularen Geometrie (z. B. dem Bindungsabstand) ab. Für diese spezifische Konfiguration und nach diesen Transformationen ergibt sich ein einfacher Qubit-Hamiltonoperator:
Hier repräsentiert den Identitätsoperator und den Pauli-Z-Operator, der auf ein einzelnes Qubit wirkt. Die Koeffizienten stammen aus den Integralen, die mit dem STO-6G-Basissatz bei diesem bestimmten Bindungsabstand und entsprechender Transformation berechnet wurden.
Mit diesem definierten Hamiltonoperator können wir nun VQE verwenden, um dessen Grundzustandsenergie zu berechnen. Es ist sinnvoll, unsere berechnete Grundzustandsenergie mit bekannten Erwartungswerten zu vergleichen. Für ein einzelnes, isoliertes Wasserstoffatom (H) beträgt die Grundzustandsenergie genau -0,5 Hartree (ohne relativistische Effekte). Berechnen wir nun die exakte Grundzustandsenergie unseres spezifischen Qubit-Hamiltonoperators, wie oben definiert, und vergleichen wir sie mit relevanten bekannten Werten.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Als nächstes benötigen wir einen parametrisierten Quantenschaltkreis — einen Ansatz — um eine Testwellenfunktion für den Grundzustand vorzubereiten. Das Ziel besteht darin, die Parameter zu finden, die den Energie-Erwartungswert minimieren. Die Wahl des Ansatzes ist entscheidend, denn sie bestimmt die Menge der möglichen Quantenzustände, die unser Schaltkreis erzeugen kann. Ein „guter" Ansatz ist flexibel genug, um einen Zustand nahe am wahren Grundzustand des untersuchten Hamiltonoperators darzustellen, aber nicht so komplex, dass er zu viele Parameter oder einen zu tiefen Schaltkreis für aktuelle Quantencomputer erfordert.
Hier werden wir drei verschiedene Ein-Qubit-Ansätze ausprobieren, um zu sehen, welcher eine bessere „Abdeckung" der möglichen Quantenzustände eines einzelnen Qubits bietet. Die „Abdeckung" bezeichnet dabei die Bandbreite der Quantenzustände, die der Ansatz-Schaltkreis durch Variation seiner Parameter erzeugen kann.
Wir verwenden drei Ansätze, die auf verschiedenen Kombinationen von Ein-Qubit-Rotationsgattern basieren:
- Ein-Achsen-Rotationsgatter-Ansatz: Dieser Ansatz verwendet Rotationen nur um eine einzige Achse (). Auf der Bloch-Kugel entspricht dies einer Bewegung entlang nur eines bestimmten Kreises. Dies ist der am wenigsten flexible Ansatz und deckt nur eine begrenzte Menge von Zuständen ab.
- Zwei-Achsen-Rotationsgatter-Ansätze: Diese Ansätze kombinieren Rotationen um zwei verschiedene Achsen ( und ). Das ermöglicht es uns, im Vergleich zur Ein-Achsen-Rotation einen größeren Teil der Bloch-Kugel zu erreichen.
Durch den Vergleich der VQE-Ergebnisse, die mit diesen drei Ansätzen erzielt werden, können wir sehen, wie die Flexibilität und die Zustandsraumabdeckung des Ansatzes unsere Fähigkeit beeinflussen, die wahre Grundzustandsenergie unseres vereinfachten Hamiltonoperators zu finden. Ein flexiblerer Ansatz hat das Potenzial, eine bessere Näherung zu finden, kann aber auch für den klassischen Optimierer schwieriger zu handhaben sein.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Nun generieren wir für jeden Parameter 5.000 Zufallszahlen und stellen die Verteilung der zufälligen Quantenzustände dar, die von den drei Ansätzen mit diesen zufälligen Parametern erzeugt werden. Du kannst dir diese Parameter wie Rotationen um verschiedene Achsen auf einer Kugeloberfläche vorstellen. Um die Verteilung der Quantenzustände zu veranschaulichen, verwenden wir die Bloch-Kugel — eine dreidimensionale Kugel, die den Zustand eines einzelnen Qubits darstellt. Jeder Punkt auf der Kugel repräsentiert einen möglichen Zustand des Qubits, wobei Nord- und Südpol dem klassischen „0" und „1" entsprechen; das Qubit kann sich jedoch auch überall dazwischen befinden und besondere Quanteneigenschaften wie Superposition zeigen. Bereite zunächst die nötigen Funktionen vor, um die 3D-Bloch-Kugel darzustellen, und generiere 5.000 zufällige Parameter.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Schauen wir uns an, wie unser erster Ansatz funktioniert.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
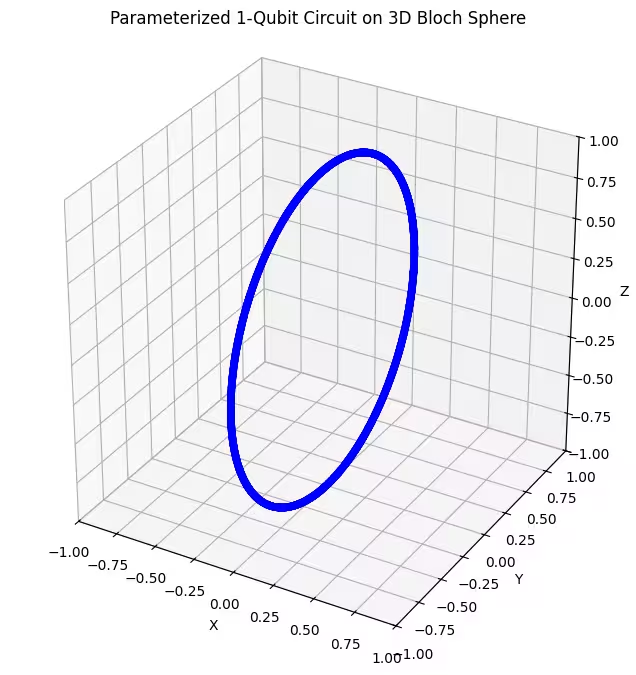
Wir sehen, dass unser erster Ansatz ringförmig verteilte Quantenzustände auf der Bloch-Kugel liefert. Das ist plausibel, denn wir haben dem Ansatz nur einen einzigen Rotationsparameter gegeben. Dadurch kann er ausschließlich Zustände erzeugen, die um eine Achse rotiert sind. Wenn man vom Punkt startet und um eine Achse dreht, ergibt sich stets ein Ring. Schauen wir uns nun unseren zweiten Ansatz an, der zwei orthogonale Rotationsgatter besitzt — Rx und Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
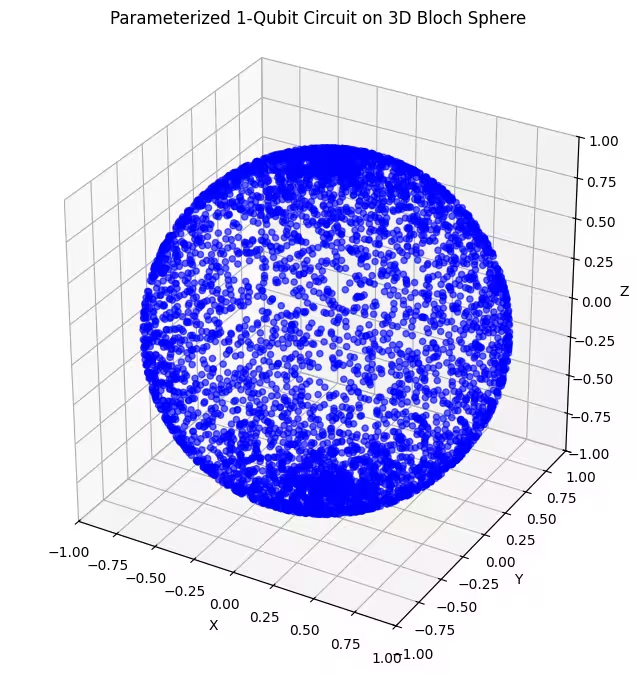
Hier sehen wir, dass unser zweiter Ansatz einen größeren Teil der Bloch-Kugel abdeckt — beachte jedoch, dass die Punkte an den Polen dichter und am Äquator stärker verstreut sind. Jetzt ist es Zeit, unseren letzten Ansatz zu überprüfen.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
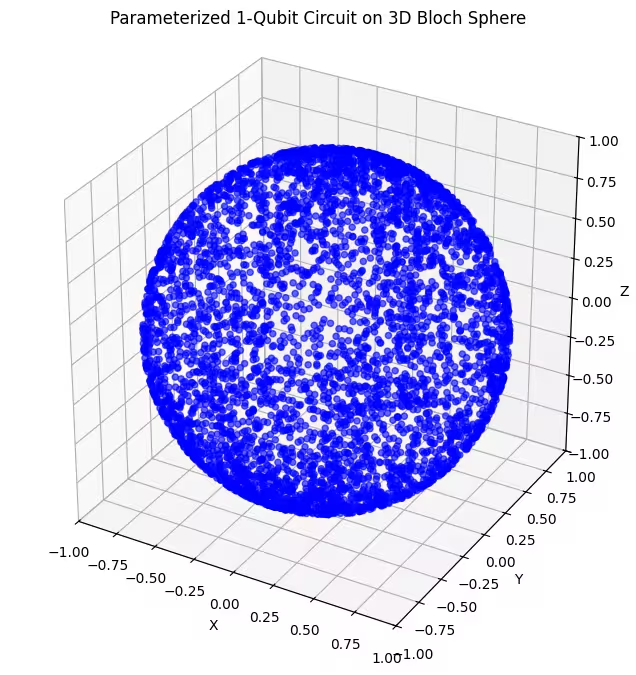
Hier sind die von unserem letzten Ansatz erzeugten Quantenzustände gleichmäßiger verteilt.
Wie bereits erwähnt, ist es am besten, Wissen über den gesuchten Grundzustand zu sammeln und einen Ansatz zu wählen, der gut geeignet ist, Zustände in der Nähe dieses Grundzustands zu erkunden. Wenn wir zum Beispiel wüssten, dass unser Grundzustand in der Nähe eines Pols liegt, würden wir Ansatz 2 wählen. Der Einfachheit halber bleiben wir bei Ansatz 3, der die gesamte Bloch-Kugel gleichmäßig abtastet.
Nachdem wir unseren Ansatz ausgewählt haben, zeichnen wir nun den Schaltkreis.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Schritt 2: Für die Zielhardware optimieren
Wenn wir eine Berechnung auf einem echten Quantencomputer durchführen, ist nicht nur die Logik des Quantenschaltkreises relevant. Es kommt auch darauf an, welche Operationen der jeweilige Quantencomputer ausführen kann und wo sich die von uns verwendeten Qubits auf dem Quantencomputer befinden — liegen sie direkt nebeneinander oder weit voneinander entfernt? Daher besteht der nächste Schritt darin, unseren Schaltkreis mit Gattern umzuschreiben, die für den verwendeten Quantencomputer native sind, und dabei das Qubit-Layout zu berücksichtigen. Dies geschieht durch transpilation — nach diesem Prozess kannst du sehen, wie unser einfacher Ansatz in einen anderen Satz von Gattern umgewandelt wird und unsere abstrakten Qubits auf physische Qubits eines echten Quantencomputers abgebildet werden.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Man sieht, dass die rx, rz-Gatter unseres Ansatzes in eine Abfolge von rz, sx-Gattern umgewandelt wurden, welche die nativen Gatter unseres Backends sind. Außerdem ist unser q0 nun auf das fünfte physische Qubit abgebildet. Wir müssen auch unseren Hamiltonoperator entsprechend dieser Änderungen anpassen, wie im folgenden Code:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Schritt 3: Auf der Zielhardware ausführen
Jetzt ist es an der Zeit, unser VQE auf einem echten QPU laufen zu lassen. Dafür brauchen wir zunächst eine Kostenfunktion für den Optimierungsprozess, die den Erwartungswert des Hamiltonoperators für einen vom Ansatz erzeugten Quantenzustand berechnet. Keine Sorge! Du musst nicht alles selbst programmieren. Wir haben eine Funktion dafür vorbereitet — du musst nur die folgende Zelle ausführen.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Abschließend bereiten wir die Anfangsparameter für unseren Ansatz und seinen Optimierungsprozess vor. Du kannst einfach lauter Nullen oder zufällige Werte verwenden. Wir haben unten bestimmte Anfangsparameter ausgewählt, du kannst aber gerne Zeilen in der Zelle kommentieren oder auskommentieren, um Parameter gleichmäßig zufällig aus dem Bereich 0 bis zu ziehen.
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Herzlichen Glückwunsch! Du hast dein erstes Quantenchemie-Experiment erfolgreich abgeschlossen. Wir sehen einen Unterschied zwischen der exakten Grundzustandsenergie des Hamiltonoperators und unserem Ergebnis, doch da wir eine Standard-Fehlerminderungstechnik (die Auslesefehler korrigiert) verwendet haben, ist die Abweichung gering. Das ist ein sehr guter Anfang!
Hinweis: Ein besseres Ergebnis lässt sich durch Einstellen einer Fehlerminderungs-Stufe über resilience_level erzielen. Der Standardwert ist 1; ein höherer Wert nutzt mehr QPU-Zeit, liefert aber möglicherweise ein besseres Ergebnis.
Schritt 4: Nachverarbeitung
Jetzt ist es an der Zeit zu betrachten, wie unser klassischer Optimierer gearbeitet hat. Führe die folgende Zelle aus und beobachte das Konvergenzmuster.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Wir sind mit einem recht guten Anfangswert gestartet und haben dadurch bereits in nur 10 Schritten einen guten Endwert erhalten. Du siehst große und kleine Ausschläge — das ist das typische Merkmal des COBYLA-Optimierers: Er durchsucht den Raum, als könnte er die Landschaft nicht sehen, und passt die Schrittweiten mit jeder Messung an.
Verständnistest
Was beobachtest du? Welcher Teil des obigen Prozesses lässt sich verbessern, um Ergebnisse näher an den theoretischen Werten oder näher an der exakten Grundzustandsenergie des Hamilton-Operators zu erhalten? Was sind dabei wichtige Überlegungen?
Antwort
Das Erste, was es zu bedenken gilt, ist die Wahl des Basissatzes, der zur Berechnung des Hamilton-Operators des Moleküls verwendet wird. Wie bereits erwähnt, beträgt die Grundzustandsenergie des H-Atoms -0,5 Hartree — ein bekannter Wert —, und der von uns gewählte STO-6G-Basissatz reicht nicht aus, um diesen Wert präzise herzuleiten.
Die Wahl eines komplexeren Basissatzes erhöht die Anzahl der vom Hamilton-Operator verwendeten Qubits; daher müssen wir für Chemieprobleme einen komplexeren und geeigneteren Ansatz auswählen.
Als Nächstes sollte das Rauschmanagement im QPU optimiert werden. Fortgeschrittenere Fehlerminderungstechniken liefern bessere Ergebnisse, können aber mehr Zeit in Anspruch nehmen. Überlege außerdem, wie sich die shot_number auf die Ergebnisse auswirkt.
Schließlich lässt sich eine bessere Konvergenzleistung auch durch das Ausprobieren verschiedener Optimierer erzielen.
Grundzustandsenergie des Wasserstoffmoleküls mit VQE berechnen
Nachdem wir den VQE-Prozess anhand einzelner -Atome betrachtet haben, berechnen wir nun die Grundzustandsenergie des -Moleküls etwas zügiger.
Schritt 1: Problem auf Quantenschaltkreise und Operatoren abbilden
Hier stellen wir dir ebenfalls einen Ein-Qubit-Hamiltonoperator bereit, der die STO-6G-Basis und die Jordan-Wigner-Transformation verwendet, mit Qubit-Reduktion durch Ausnutzung einer Symmetrie des Hamiltonoperators. Beachte, dass wir einen Atomabstand von 0.735 zwischen den beiden Wasserstoffatomen verwendet haben.
Im Unterschied zur Berechnung eines einzelnen Wasserstoffatoms () müssen wir für die Grundzustandsberechnung des Wasserstoffmoleküls () zusätzlich zur Energie der Elektronenorbitale auch die Abstoßungskraft zwischen den Atomkernen der beiden Wasserstoffatome berücksichtigen. In diesem Schritt geben wir diesen Wert als Konstante vor; in der Verständnisfrage wirst du ihn selbst berechnen.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Schritt 2: Für die Zielhardware optimieren
Da die Anzahl der Qubits des vorherigen VQE und des Hamiltonoperators mit der des zu verwendenden Backends übereinstimmt, nutzen wir den bestehenden Ansatz und seine optimierte Form.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Schritt 3: Auf der Zielhardware ausführen
Jetzt ist es an der Zeit, die Berechnungen auf dem echten QPU durchzuführen. Fast alles ist gleich, aber wir wählen einen geeigneten Startpunkt für den Hamiltonoperator. Außerdem werden einige Einstellungen des Estimator, der zur Berechnung der Erwartungswerte des Hamiltonoperators für den Ansatz auf dem QPU verwendet wird, im iterativen Teil etwas anders gesetzt als bei den vorherigen Berechnungen. Diese Änderung besprechen wir in einer Verständnisfrage.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Obwohl VQE theoretisch eine obere Schranke für die wahre Grundzustandsenergie liefert, können praktische Implementierungen auf echter oder verrauschter simulierter Quantenhardware sowie Näherungen bei der Vorbereitung des Hamiltonoperators (wie Basissätze oder Qubit-Reduktion) Fehler einführen, die gelegentlich zu einem gemessenen Energiewert führen, der leicht unterhalb des exakten theoretischen Wertes oder eines bestimmten numerischen Referenzwertes liegt. Obwohl also gewisse Fehler vorhanden sind, erscheinen die Ergebnisse zufriedenstellend – besonders angesichts der geringen Anzahl an Schritten. Schauen wir uns nun abschließend an, wie der Optimierer gearbeitet hat.
Schritt 4: Nachverarbeitung
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Verständnis überprüfen
Berechne die Kernabstoßungsenergie des -Moleküls, die wir oben als Konstante (0.71997 Hartree) angegeben haben.

Verwende das Coulombsche Gesetz und atomare Einheiten, um das Ergebnis in Hartree zu erhalten.
Antwort
Da beide Wasserstoffkerne positiv geladen sind, stoßen sie sich aufgrund der elektrostatischen Kraft ab. Diese Abstoßung wird durch das Coulombsche Gesetz beschrieben:
wobei die Ladung des Protons, die elektrische Feldkonstante (Permittivität des Vakuums) und der Abstand zwischen den beiden Kernen in Metern oder Bohr-Radien (Einheit: Joule (J)) ist.
Um diese Energie in Hartree zu berechnen, müssen wir die obige Gleichung in das System der Atomaren Einheiten (AE) überführen. In AE gilt: , und der Bohr-Radius () ist 1 und bildet die grundlegende Längeneinheit. Mit diesen Vereinfachungen reduziert sich das Coulombsche Gesetz auf:
wobei in Bohr-Radien () gemessen werden muss.
Um den gegebenen Kernabstand von in umzurechnen, benötigen wir die folgende Umrechnungsbeziehung:
also wird zu .
Die Kernabstoßungsenergie des gegebenen beträgt damit:
Reaktionsenergie von berechnen
Jetzt nutzen wir die gewonnenen Ergebnisse! Du hast VQE, den variationellen Quanteneigenwertlöser, verwendet, um die Grundzustandsenergie des -Atoms und des -Moleküls zu berechnen. Was noch fehlt, ist die Nutzung dieser Werte zur Berechnung der Reaktionsenergie des Prozesses .
Die Reaktionsenergie ist die Energieänderung, die auftritt, wenn Stoffe miteinander reagieren und neue Stoffe bilden. Stell dir vor, du baust etwas: Manchmal musst du Energie hineinstecken (wie beim Stapeln von Blöcken), manchmal wird Energie freigesetzt (wie eine Kugel, die einen Hügel hinunterrollt). In der Chemie nehmen Reaktionen entweder Energie auf (endotherm) oder setzen Energie frei (exotherm).
Die Reaktionsenergie des Prozesses lässt sich mit folgender Formel berechnen:
Führe die folgende Zelle aus, um dies grafisch darzustellen. Wir verwenden hier den exakten Grundzustandswert jedes Hamiltonoperators und vergleichen die Reaktionsenergie der exakten Lösung mit den VQE-Ergebnissen.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Wie die Abbildung zeigt, stimmen die exakten Grundzustandsenergien der Hamiltonoperatoren und die mit den VQE-Ergebnissen berechnete Reaktionsenergie trotz gewisser Fehler gut überein und liegen beide nahe bei -0,2 Hartree.
Es ist an dieser Stelle wichtig festzuhalten, dass die Reaktionsenergie dieses Prozesses einen negativen Wert hat. Das bedeutet, dass beim Prozess Energie freigesetzt wird und das entstehende Molekül eine niedrigere Energie besitzt als zwei einzelne Atome.
Fassen wir zusammen, was wir bislang gelernt haben.
Zunächst haben wir uns zwei wichtige Näherungsverfahren angeschaut, die zur Lösung quantenchemischer Probleme benötigt werden: das Variationsprinzip und die Wahl des Basissatzes – beides sind grundlegende Bestandteile von VQE. Wir haben das Variationsprinzip selbst angewendet und die Grundzustandsenergie des harmonischen Oszillators von Hand berechnet.
Anschließend haben wir VQE erkundet, einen weit verbreiteten Algorithmus zur Berechnung der Grundzustandsenergie eines Quantensystems. Wir haben Code ausgeführt, um die Grundzustandsenergien des Wasserstoffatoms () und des Wasserstoffmoleküls () zu berechnen. Dabei haben wir insbesondere gelernt, dass es notwendig ist, den geeigneten molekularen Hamiltonoperator für das System zu ermitteln und ihn in eine auf einem Quantencomputer ausführbare Form zu überführen. Wir haben außerdem gesehen, dass der Ansatz – ein parametrisierter Quantenschaltkreis – benötigt wird, um Versuchs-Quantenzustände im Rahmen von VQE vorzubereiten, und die Bedeutung der Wahl einer geeigneten Ansatz-Schaltkreisstruktur diskutiert. Zudem haben wir gelernt, dass VQE auf einem iterativen Optimierungsprozess mit einem klassischen Computer basiert, der den Quantenschaltkreis auf den Zustand niedrigster Energie hinführt, und konnten die Konvergenz dieses Prozesses beobachten.
Schließlich haben wir die durch VQE berechneten Grundzustandsenergien von und genutzt, um die Reaktionsenergie des Prozesses zu bestimmen.
VQE ist ein leistungsstarker Quantenalgorithmus für die nahe Zukunft (NISQ-Ära), aber es ist wichtig, seine Grenzen zu kennen. Die Leistung von VQE hängt stark von der Wahl des Ansatzes ab – es wird zunehmend schwierig, einen effizient präparierbaren Ansatz zu finden, der den wahren Grundzustand für größere, komplexere Moleküle gut beschreibt. Darüber hinaus ist aktuelle Quantenhardware anfällig für Rauschen, was die Genauigkeit der VQE-Ergebnisse beeinträchtigen kann, insbesondere bei tieferen Schaltkreisen oder einer größeren Anzahl von Qubits. Trotz dieser Herausforderungen ist VQE ein grundlegender Algorithmus, und laufende Forschung untersucht ausgefeiltere Variationsmethoden sowie Fehlerminderungstechniken, um die Grenzen des in der Quantenchemie auf naher Quantenhardware Machbaren zu verschieben. So werden beispielsweise Algorithmen wie Sample-based Quantum Diagonalization (SQD) entwickelt, die Stichproben aus Quantenschaltkreisen mit klassischer Diagonalisierung in einem Unterraum kombinieren, um die Energieschätzung zu verbessern und einige der Einschränkungen von VQE – insbesondere hinsichtlich Messeffizienz und Robustheit gegenüber Rauschen – zu adressieren.
Wiederholung und Fragen
Wichtige Konzepte:
- Ein variationeller Quantenalgorithmus ist ein Rechenparadigma, bei dem ein klassischer Computer und ein Quantencomputer zusammenarbeiten, um ein Problem zu lösen.
- Bei VQE beginnen wir mit dem Hamiltonoperator unseres Systems und bilden ihn auf Qubits ab, um ihn auf dem Quantencomputer auszuführen. Wir wählen einen parametrisierten Quantenschaltkreis (einen Ansatz) und führen wiederholte Messungen durch, wobei wir die Parameter des Ansatzes variieren, bis der niedrigste Energiewert erreicht ist. Die Suche im Parameterraum erfolgt mit einem klassischen Optimierer. Um gute Ergebnisse zu erzielen, ist es notwendig, einen guten Ansatz und einen geeigneten Optimierer zu wählen.
- Die Reaktionsenergie ist die gesamte Energieänderung bei einer chemischen Reaktion und wird durch die Differenz zwischen der Energie der Reaktanten und der Produkte bestimmt.
Wahr/Falsch
- Das Variationsprinzip besagt, dass der Erwartungswert der Energie für jede Versuchs-Wellenfunktion stets größer oder gleich der wahren Grundzustandsenergie ist.
- Ein Basissatz ist eine Sammlung von Funktionen, die zur Näherung von Quantenwellenfunktionen verwendet werden.
- VQE ist ein Quantenalgorithmus, der die Schrödinger-Gleichung für einen gegebenen Hamiltonoperator exakt löst.
- Bei VQE wird ein parametrisierter Quantenschaltkreis (ein Ansatz) verwendet, um Versuchs-Wellenfunktionen vorzubereiten.
- Die Wahl des Optimierers in VQE (zum Beispiel COBYLA, SPSA oder ADAM) hat keinen Einfluss auf die Qualität des Ergebnisses.
- Qiskits
Estimatorwird verwendet, um Erwartungswerte von Hamiltonoperatoren in VQE direkt zu berechnen.
Multiple-Choice-Fragen:
- Welche Rolle spielt der Hamiltonoperator in VQE?
- A) Zufällige Quantenzustände erzeugen
- B) Die Energie von Quantenzuständen bestimmen
- C) Quantenschaltkreise optimieren
- D) Verschränkung erzeugen
- Was ist das primäre Ziel des VQE-Algorithmus?
- A) Die Grundzustandsenergie eines Hamiltonoperators zu finden
- B) Verschränkung zwischen Qubits herzustellen
- C) Grovers Suchalgorithmus durchzuführen
- D) Die RSA-Verschlüsselung zu brechen
- Wie viele Quantenzustände werden in diesem Notebook erzeugt, um den Ansatz zu vergleichen?
- A) 100
- B) 1000
- C) 5000
- D) 10.000
- Warum wird in VQE ein klassischer Optimierer benötigt?
- A) Um Quantenmessungen durchzuführen
- B) Um die Ansatz-Parameter zu aktualisieren und die Energie zu minimieren
- C) Um Qubits zu verschränken
- D) Um Quanten-Zufälligkeit zu erzeugen
- Warum ist der Ansatz so konzipiert, dass er parametrisiert ist?
- A) Um die Vorbereitung von Quantenzuständen zu ermöglichen
- B) Um einen weiten Raum von Quantenzuständen durchsuchen zu können
- C) Um die Schaltkreiskomplexität zu reduzieren
- D) Um Eigenwerte direkt zu messen
- Welche der folgenden Aussagen trifft am besten auf die Wahl eines guten Ansatzes zu?
- A) Ein Ansatz muss Zustände gleichmäßig auf der Bloch-Kugel verteilen, sonst schlägt er fehl.
- B) Ein Ansatz sollte auf das jeweilige System zugeschnitten sein, damit er Zustände nahe dem Grundzustand erzeugen kann.
- C) Ein Ansatz sollte mit seinen variationellen Parametern zufällige Zustände erzeugen.
- D) Ein besserer Ansatz hat immer mehr variationelle Parameter.
(Optional) Anhang: Optimierer-Overhead nach Ansatz-Komplexität
VQE ist mit einigen bekannten Herausforderungen[ref 6] konfrontiert, von denen die folgenden mit dem bisher Gelernten zusammenhängen.
- Herausforderungen bei der Ansatz-Wahl
Die Wahl des richtigen variationellen Ansatzes ist von Natur aus schwierig. Chemisch inspirierte Ansätze (wie UCCSD) liefern physikalische Genauigkeit, erfordern aber tiefe Schaltkreise, während hardwareeffiziente Ansätze flachere Schaltkreise haben, dafür aber möglicherweise an physikalischer Interpretierbarkeit einbüßen. Zudem führen viele Ansätze übermäßig viele variationelle Parameter ein, die kaum zur Verbesserung der Genauigkeit beitragen, die Optimierung aber erheblich erschweren.
- Schwierigkeiten bei der Optimierung
Die Optimierungslandschaft von VQE kann Bereiche aufweisen, in denen Gradienten exponentiell verschwinden (sogenannte Barren Plateaus), was es klassischen Optimierern erschwert, die variationellen Parameter effizient zu aktualisieren. Forscher haben daher versucht, verschiedene Optimierer-Typen einzusetzen – gradientenbasierte und gradientenfreie –, aber beide stoßen auf Schwierigkeiten. Gradientenbasierte Optimierer leiden unter Barren Plateaus, während gradientenfreie Methoden eine große Anzahl von Funktionsauswertungen benötigen.
- Optimierer-Overhead
Eine weitere bekannte Herausforderung ist der Optimierer-Overhead, der mit der Skalierung des Problems zusammenhängt. Die für VQE benötigten Quantenschaltkreise wachsen in Tiefe und Komplexität mit zunehmender Problemgröße; dies erhöht in der Regel auch die Anzahl der zu optimierenden Parameter. Der Optimierungsprozess wird schwer handhabbar, wenn die Anzahl der Parameter zunimmt, was zu langsamer Konvergenz und Schwierigkeiten beim Finden der optimalen Lösung führt.
Im Folgenden werfen wir einen Blick auf diese Herausforderungen, indem wir VQE für ein -Molekül mit zwei verschiedenen Ansatz-Typen verwenden.
(Hinweis: Dies kann mehr QPU-Zeit in Anspruch nehmen – verwende gerne einen Simulator, falls du nicht genug Zeit hast.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Jetzt starten wir einen VQE mit einem Startpunkt aus lauter Einsen, einem Maximum von 20 Schritten, und vergleichen die Konvergenz beider Läufe.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Die obige Grafik zeigt deutlich, dass der Optimierungsprozess des Ansatzes mit mehr Variablen länger braucht, um eine stabile Konvergenz zu erreichen.
Statt sich auf einfache Ein-Qubit-Schaltkreise und einen unkomplizierten Ansatz zu verlassen, steigt die Komplexität der Optimierung, wenn größere Quantenschaltkreise und komplexer strukturierte Ansätze erforderlich sind. Dies verdeutlicht eine bekannte Herausforderung bei VQEs: den Overhead des Optimierers.
Forscher entwickeln weiterhin verschiedene fortschrittliche Methoden, die Quantencomputer für Chemieprobleme nutzen können. Eine Vielzahl von Lernmaterialien findest du auf IBM Quantum Learning.
Referenzen
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)